Multi-resistance strategy for viral diseases and in vitro short hairpin RNA verification method in pigs
Article information

Abstract
Objective
Foot and mouth disease (FMD) and porcine reproductive and respiratory syndrome (PRRS) are major diseases that interrupt porcine production. Because they are viral diseases, vaccinations are of only limited effectiveness in preventing outbreaks. To establish an alternative multi-resistant strategy against FMD virus (FMDV) and PRRS virus (PRRSV), the present study introduced two genetic modification techniques to porcine cells.
Methods
First, cluster of differentiation 163 (CD163), the PRRSV viral receptor, was edited with the clustered regularly interspaced short palindromic repeats-CRISPR-associated protein 9 technique. The CD163 gene sequences of edited cells and control cells differed. Second, short hairpin RNA (shRNAs) were integrated into the cells. The shRNAs, targeting the 3D gene of FMDV and the open reading frame 7 (ORF7) gene of PRRSV, were transferred into fibroblasts. We also developed an in vitro shRNA verification method with a target gene expression vector.
Results
shRNA activity was confirmed in vitro with vectors that expressed the 3D and ORF7 genes in the cells. Cells containing shRNAs showed lower transcript levels than cells with only the expression vectors. The shRNAs were integrated into CD163-edited cells to combine the two techniques, and the viral genes were suppressed in these cells.
Conclusion
We established a multi-resistant strategy against viral diseases and an in vitro shRNA verification method.
INTRODUCTION
Contagious diseases must be controlled at animal farms to maintain domestic animal productivity as diseases can cause delayed growth and even death in farm animals. Foot and mouth disease (FMD) and porcine reproductive and respiratory syndrome (PRRS) are significant viral diseases that occur on pig farms and are spread by air. FMD is a one of the most widespread problems in cloven-hoofed animals; it causes fever and sores on the foot and mouth and has high mortality rates [1,2]. PRRS outbreaks cause huge economic damage in the pig industry because of reproductive failure and disorders of the respiratory tract [3,4]. Damage to the animals and farms is serious and broad in economic and biological terms [5]. Thus, it is more efficacious to block viral infection rather than care for infected animals. However, the genetic diversity among serotypes has made it difficult to understand these viral diseases. Although many FMD virus (FMDV) and PRRSV virus (PRRSV) vaccines have been developed, not all serotypes of the viruses are included [6,7]. For this reason, the alternative solution of producing genetically modified animals with disease-resistance has been examined by several researchers.
An approach to producing viral disease-resistant animals is knockout of the receptors involved in viral infection. Several receptors associated with virus binding, internalization, and uncoating have been identified [8]. Whitworth et al produced cluster of differentiation 163 (CD163)-knockout pigs which are resistant to PRRSV (CD163; the receptor of PRRSV) [9,10]. Many researchers have tried to suppress viral replication using the RNA interference (RNAi) technique. The 3D gene, a viral RNA polymerase in FMDV, has a vital role in virus replication [11]. Virus replication has been successfully repressed by inhibiting transcription through RNA polymerase knockdown using an RNAi system. PRRSV replication can be significantly suppressed by interrupting formation of the open reading frame 7 (ORF7) gene and organizing nucleocapsid protein N [12–14]. Interestingly, because DNA sequences of these target genes are highly conserved among serotypes due to their pivotal roles [15,16], carefully designed RNAi vectors could be applied to target various viral serotypes.
Despite the many significant results, previous studies have focused on targeting a virus using a single technique. Although a challenge test with animals is required to test the efficiency of RNAi, animal testing using live viruses is costly and time-consuming. Therefore, an alternative in vitro RNAi verification method is important as a check on the overall experiment. In this study, we used two disease-resistance techniques to produce a multi-resistant pig and developed an in vitro RNAi verification method. A CD163 was knocked out, and knockdown vectors targeting viral genes including 3D and ORF7 were introduced into pig somatic cells. The in vitro verification was applied to confirm short hairpin RNA (shRNA) activity in the cells. Both shRNAs were integrated into CD163-knockout cell lines, and shRNA activity in these multi-resistant cells was verified.
MATERIALS AND METHODS
Animal care
The care and experimental use of pigs were approved by the Institutional Animal Care and Use Committee at Seoul National University (Approval No.: SNU-140328-2). A pregnant sow was purchased from an animal farm. The sow was taken care of exclusively by the farm and sacrificed 30 days after artificial insemination at a slaughterhouse (Hanbo, Gyeongsangnamdo, Korea) under approval by the Korean government.
Design of CRISPR-Cas9 vector targeting pig CD163
The clustered regularly interspaced short palindromic repeats-CRISPR-associated protein 9 (CRISPR-Cas9) system was used to knockout CD163 in pig fetal fibroblasts. The single guide RNA (sgRNA) target sequence against pig CD163 (Gene ID: 397031) was determined with an online search tool (https://www.atum.bio) (Figure 1A). To reduce the off-target effects of CRISPR-Cas9, various sgRNA target sequences were aligned with porcine whole genome data using the BLAST program (National Center for Biotechnology Information [NCBI] web site, https://blast.ncbi.nlm.nih.gov/BlastAlign.cgi). Finally, DNA constructs carrying the target sequence were designed by adding PAM and a guide sequence (oligonucleotide sequences in Table 1). Pairs of oligonucleotides were dimerized by slow cooling from 95°C to 25°C (5°C/min) and then inserted into the pSpCas9(BB)-2A-GFP (PX458; Addgene, Cambridge, MA, USA) plasmid vector according to the manufacturer’s instructions (T4 DNA ligase; Invitrogen, Carlsbad, CA, USA). The recombinant plasmid vector was transformed into Escherichia coli (DH5α; Novagen, Wilmington, DE, USA). The purified plasmid vectors from E. coli were introduced into pig fetal fibroblasts by lipofection, as described below.

Target sequences of shRNA and sgRNA and overall scheme of experiment. (A) sgRNA target sequences for the CD163 gene. (B) shRNA sequences against the 3D gene of foot and mouth disease virus and ORF7 gene of porcine reproductive and respiratory syndrome virus. Underlined sequences are target sequences. (C) Brief process of CD163 knockout (Dark grey for target 1 and light grey for target 2); (D) shRNA introduction; (E) shRNA transduction into CD163-knockout cells. shRNA, short hairpin RNA; sgRNA, single guide RNA; CD163, cluster of differentiation 163; ORF, open reading frame.

Oligonucleotides for single guide RNA
Production of a lentiviral vector carrying shRNA
The genomic sequences of three FMDV genes and three PRRSV genes from different serotypes were aligned to design the shRNAs against various subtypes of the FMD and PRRS viruses (Gene ID numbers and base numbers in Table 2). Based on the aligned sequences, shRNA targeting part of the conserved sequences was selected using an online tool (https://www.thermofisher.com) (Figure 1B). The shRNA sequences were aligned to the whole porcine transcriptome to prevent the unintended binding of shRNA on the pig genome (taxid: 9823) through the BLAST program (NCBI web site). Finally, the DNA constructs harboring shRNA sequences were designed by adding a nine-nucleotide loop (TTCAAGAGA) and an Xho I restriction site (oligonucleotide sequences in Table 3).

Gene ID of viral genes (nucleotide base numbers for complete genome)

Oligonucleotides for shRNA
Pairs of oligonucleotides were dimerized by slow cooling from 95°C to 4°C (1°C/min) and then inserted into the pLL3.7 plasmid vector according to the manufacturer’s instructions. Lentiviral vectors were produced using these recombinant plasmid vectors as described previously [17]. In brief, HEK 293 LTV cells (Cell Biolabs, San Diego, CA USA) were used as packaging cells. Four plasmids (pLL3.7, pLP1, pLP2, and pLP/VSVG; Invitrogen, USA) were transfected into HEK 293 LTV cells using the calcium phosphate precipitation method. After 36 h, the culture supernatants were harvested four times (every 12 h) and then concentrated by centrifugation at 18,000× g for 5 h at 4°C. The virus pellet was dissolved in Dulbecco’s modified Eagle’s medium (DMEM) and stored at −80°C until use.
Lipofection into pig fetal fibroblasts and viral infection
Pig fetal fibroblasts (pFFs) from a 30-day porcine fetus were used to establish viral disease-resistant cell lines. The pFFs were cultured with DMEM (low glucose; Welgene, Daegu, Korea) supplemented with 10% fetal bovine serum, 1× glutamax, 0.1 mM β-mercaptoethanol, and 1× antibiotic-antimycotic (Gibco, Grand Island, NY, USA). Cells reaching confluence were subcultured into a new culture plate by dissociating with 0.04% trypsin/ethylenediaminetetraacetic acid solution (Welgene, Korea).
The recombinant PX458 vectors were introduced into pFFs using the Lipofectamine 3000 Transfection Reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions (Figure 1C). The cells were diluted and seeded into 96-well dishes at a concentration of one cell per well 1 week post-lipofection. Colonies from a single cell were cultured for over 2 weeks, and genomic DNA (gDNA) was extracted from single cultured pFF cells using the G-spin Total DNA Extraction Mini Kit (Intron Biotech, Sungnam, Korea). The target region in pig CD163 was amplified with specific primers (Table 4), and amplicons were sequenced using an ABI PRISM 3730 automated sequencer (Applied Biosystems, Foster City, CA, USA) to identify mutations using CRISPR-cas9. Cultured pFFs were infected with lentiviral vectors harboring shRNAs for 24 h. After another 5 to 7 days, the shRNA functions in the infected cells were analyzed as described below (Figure 1D). Finally, to establish multi-disease-resistant cell lines, we introduced shRNAs into a CD163-knockout cell line by lentiviral vectors as mentioned above (Figure 1E).

Polymerase chain reaction primers
Verification of shRNA knockdown efficiency against FMD and PRRS viral genes
Oligonucleotides carrying partial sequences of the viral genes (Genbank ID of 3D; KF694745.1, ORF7; AY585241.1) were cloned into the pcDNA3-enhanced green fluorescent protein (EGFP) expression vector (Addgene, USA). Plasmid vectors expressing viral genes were transfected into shRNA-expressed pFFs with various concentrations using Lipofectamine 3000 Transfection Reagent (Thermo Fisher Scientific, USA). After 48 h, the total RNA of the transfected pFFs was isolated with TRIzol Reagent (Invitrogen, USA) according to the manufacturer’s manual, and cDNA was synthesized with a High-Capacity RNA-to-cDNA Kit (Applied Biosystems, USA) following the standard protocol. mRNA levels of the viral genes were analyzed by real-time polymerase chain reaction (PCR) with the primers shown in Table 4; then, the relative mRNA level was measured by normalizing the threshold cycle (Ct) values of each gene to that of the beta-actin [18]. The mRNA levels of the viral genes in the multi-resistant cell lines were measured using the same method.
Statistical analysis
Statistical analysis of the quantitative PCR data was performed using GraphPad Prism Software (version 5.01; San Diego, CA, USA). Significant differences among experimental groups were determined by one-way analysis of variance followed by Tukey’s multiple comparison test. A p-value <0.05 was considered significant. Data are presented as the mean± the standard error.
RESULTS
Knockout of the CD163 gene in the porcine genome
The approach to generate a pig resistant to multiple viruses involved protection against viral infection and inhibition of infected viruses. To protect against viral infection, we found receptors involved in the PRRSV infection process and performed gene knockout; RNAi technology was used to knockdown viral genes and inhibit the virus. First, CD163, known as the receptor for uncoating viral particles, was selected. Several receptors are involved in the PPRSV infectious process, such as virus binding, internalization, and uncoating [8]. A previous study showed that CD163−/− pigs do not develop any infectious symptoms, including respiratory signs, fever, or edema in lung tissue, and no viral nucleotides are detected in their blood [10]. Thus, we employed the CRISPR-Cas9 system to knockout CD163 in pFFs [9]. It was important to design a sgRNA sequence with high specificity for the target gene to minimize the off-target effect of the CRISPR-Cas9 treatment. Several target sites were selected in the CD163 sequence. Of these, candidate sequences with high scores were aligned with the whole porcine genome, and two candidates with high affinity for only CD163 were selected (Figure 2A). These candidates were cloned into the PX458 plasmid, and the vectors were introduced into pFFs using lipofection. After single cell cloning of transduced pFFs, several cell colonies with typical fibroblast morphology were selected. Finally, three cell lines were established and maintained during an extended culture period (Figure 2B). The sequencing results showed that CRISPR-Cas9 was successful on target 1, but not on target 2 (Figure 2C).
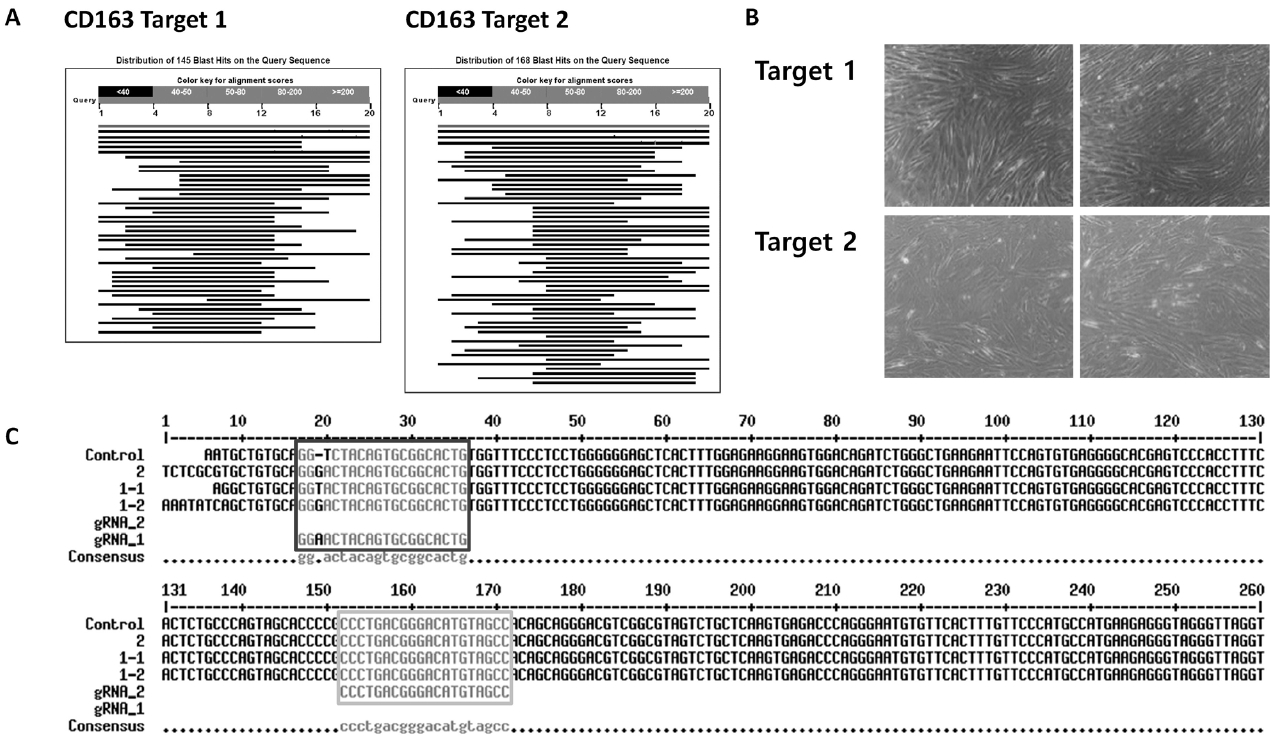
Establishment of homologous CD163-edited cells. (A) BLAST result of sgRNA target sequence with the porcine whole genome. (B) Normal status of CRISPR-Cas9-treated cell lines from single cell culture. (C) Sequencing result of control and CRISPR-Cas9-treated cell lines (Dark grey for target 1 and light grey for target 2). CD163, cluster of differentiation 163; sgRNA, single guide RNA; CRISPR-Cas9, clustered regularly interspaced short palindromic repeats-CRISPR-associated protein 9.
Transduction of shRNAs that against viral genes
We used RNAi to inhibit translation of viral genes and suppress viruses already infected into host. RNAi is a post-transcriptional regulator that controls gene expression by degrading mRNA by binding microRNA [19,20]. The shRNA is a sort of artificial microRNA that can silence target genes via the RNAi process [21]. We selected candidate genes that were highly conserved among various virus serotypes using this technique; we, in turn, produced knockdown vectors carrying shRNAs against the viral genes. It has been reported that various serotypes (FMDV: type O, A, and Asia1 [22,23]; PRRSV: type North America1, North America2, and Korean [24,25]) are widespread in Korea and Asia. We selected the viral genes that were highly conserved among viral serotypes existing in Korea and Asia. It has bfrom the various serotypes, the 3D gene of FMDV and the ORF7 of PRRSV represented the highest homology ollected the nucleotide sequences of the conserved genes from different serotypes and the NCBI ID number as described in Table 2. Among the sequences from the various serotypes, the 3D gene of FMDV and the ORF7 of PRRSV represented the highest homology (Figure 3, Table 5). Consequently, the 3D gene of FMDV and the ORF7 gene of PRRSV were chosen as knockdown target genes. Based on sequence alignment data, shRNA expression vectors were designed by considering a highly conserved region of genes among serotypes. The targeting sequences were not similar to that of the porcine transcriptome (Figure 4A). Finally, these vectors were introduced into pFFs by lentiviral transduction.

Sequence alignment of different serotypes. Alignment of foot and mouth disease virus serotypes, type O, type A, and type Asia 1 for VP1 gene (A); 2B gene (B); and 3D gene (C). Alignment of porcine reproductive and respiratory syndrome virus serotypes, North America 1, North America 2, and the Korean isolate for the ORF5 gene (D); ORF6 gene (E); and ORF7 gene (F). Black character for conserved nucleotides, dark grey for similar nucleotide, and light grey for different nucleotides. ORF, open reading frame.

Similarity and function of viral genes

Viral transfer of shRNA sequence against foot and mouth disease virus and porcine reproductive and respiratory syndrome virus. (A) BLAST result between shRNA target site and porcine transcriptome. Black indicates very low similarity. (B) Confirmation of viral gene expression from vectors. (C) Level of viral gene expressed from transfected vectors. Error bars indicate the standard error of the mean (SEM), and * indicates a significant difference. (E) Relative mRNA level of the 3D gene in CD163-edited cells. (F) Relative mRNA level of the ORF7 gene in CD163-edited cells. Error bars represent mean SEM, and * indicates a significant difference. FP, cells with both shRNAs for 3D and ORF7; f, 3D gene expression vector; p, ORF7 expression vector; fp, both vectors above; CD163, cluster of differentiation 163; ORF, open reading frame.
Development of multi-disease-resistant strategies by combining two approaches
Finally, to produce disease-resistant cells against multiple viruses, shRNAs against the 3D and ORF7 genes were transferred by lentiviral vectors into CD163-knockout pFFs. Because FMDV and PRRSV have crucial simultaneous impacts on animal farms across the country, it is important to protect against these two contagious diseases simultaneously. Three samples, including shRNA against 3D, shRNA against ORF7, and both shRNAs-treated groups, were prepared.
Establishment of in vitro shRNA verification method
Infection with live viruses is required to verify disease resistance. However, it is difficult to conduct an experiment using contagious viruses, because biohazard facilities and equipments approved by the government are required. Thus, we developed a novel method to indirectly demonstrate shRNA functions using a recombinant viral gene expression plasmid vector. The synthesized DNA constructs carrying viral genes were inserted into the pcDNA3-EGFP vector to analyze the activity and effectiveness of the shRNAs in vitro. Plasmid vectors that expressed the 3D and ORF7 genes were transfected into shRNA-expressed cells. Viral gene expression in pFFs was confirmed by PCR (Figure 4B). No significant difference was observed between the control and 1× treatment group in relative quantity of viral mRNA. The 3D and ORF7 mRNA levels decreased significantly in the shRNA treatment groups compared with the control group when the former groups were treated with 10× plasmid concentration (about 9-fold in 3D and 160-fold in ORF7; Figure 4C, 4D). These results show that the viral genes were successfully suppressed by shRNAs integrated into the pFF genome. The mRNA levels of the viral genes decreased in response to the targeted shRNAs transfected into CD163-knockout cells. Additionally, expression of the viral genes was significantly inhibited (>4.8-fold in 3D and 3.8-fold in ORF7; Figure 4E, 4F) in cells infected with both shRNAs against 3D and OFR7. Taken together, these data show that targeting efficiency of shRNAs against FMDV and PRRSV was confirmed in vitro.
DISCUSSION
A multi-disease-resistant strategy is achievable using various techniques simultaneously
Due to their genetic diversity, it is difficult to prevent viral infectious diseases [32]. Indeed, there are different nucleotide and polypeptide sequences not only across serotypes but also between subtypes in a serotype. This variety gives resistance against vaccination of the host to the viruses. Thus, a vaccination for each new type of virus is needed every time to ensure protection. Genetic modifications have been used to produce viral disease-resistant farm animals to overcome the obstacles of vaccination.
When viruses infect host cells, they recognize specific molecules on the cell surface. Heparan sulfate, sialoadhesin, and CD163 are major mediators of PRRSV infection [33]. The CD163 receptor is more strongly related to viral susceptibility than sialoadhesin according to an experimental study [34]. CD163 is a transmembrane glycoprotein on monocytes and macrophages that has been used as the subject of the PRRSV resistance experiments. Cells were treated with antibodies targeting CD163, and they suppressed entry of the virus into the cells [35,36]. CRISPR-Cas9 gene editing was conducted with porcine embryos in CD163-knockout studies. These researchers produced piglets by embryo transfer, and the pigs showed total viral resistance [9,10]. Accordingly, targeting CD163 is sufficient to block viral infection. We conducted gene editing on the fore part of the CD163 gene. Modification of the genomic sequence was detected in the clone cell lines. However, additional experiments are needed to confirm whether the polypeptide sequence or the protein structure changed.
Animals are usually vaccinated against FMDV with live vaccines [31]. As a genetic modification to deliver resistance, many attempts have been made to suppress the virus with RNAi [37]. The 3D gene is a major target for the induction of resistance. The 3D gene encodes viral RNA polymerase, which is the most important protein in the virus life cycle [16]. RNAs synthesized by viral polymerase produce other viral proteins during replication and transcribe the genetic information of the virus, especially RNA viruses, such as FMDV. Researchers have effectively decreased the replication of FMDV by miRNAs or shRNAs against the 3D gene [38,39]. Nucleocapsid protein knockdown has been conducted to suppress PRRSV. One candidate is ORF7, which encodes an essential protein. ORF7 is involved in a nucleocapsid protein N, which is a structural protein of the virus. This protein leads to encapsulation of PRRSV. Many studies have shown that interrupting encapsulation by suppressing the ORF7 gene reduces PRRSV replication [40–42]. The 3D and ORF7 genes are highly conserved among serotypes because of their importance [43,44]. Thus, shRNAs that restrict those genes can be a powerful barrier to block viral infection. The 3D and ORF7 sequences from common serotypes were aligned together, and conservation of these genes was confirmed in this study. The shRNAs that targeted 3D and ORF7 were integrated into the porcine genome.
Animals with resistance to one disease are vulnerable to invasion by other diseases. To prevent diverse diseases, animals need multi-resistance against diseases that occur frequently. In our study, the gene edition of the viral receptor and viral gene targeting shRNAs were used together (shRNAs against 3D and ORF7 were integrated into the genome of CD163 edited cells). Knockdown of 3D and ORF7 was also effective in those cells.
Efficiency of shRNA against contagious viruses was verified in vitro
A common method for ensuring the effectiveness of shRNAs against viral diseases is infecting transgenic animals that carry shRNAs. In previous studies, RNAi transgenic pigs had no complications with stable siRNA expression [45], and this is the best way to confirm the efficiency of the RNAi. However, the production of animals expressing RNAi requires infrastructures, money, and time. Even after the animals are successfully produced, handling highly infectious viruses is very dangerous. Therefore, a specialized facility is needed to control hazardous factors during experiments. In this study, we suggested an alternative method to supplement the virus challenge test. The ORF7 gene of PRRSV and the 3D gene of FMDV were expressed in recipient cells after transfection, indicating recapitulation in virus-infected cells. Our results show that the transduced shRNAs induced degradation of RNAs encoding virus proteins. This technique is easier and far less hazardous than an infection experiment. Although this method cannot completely replace animal tests, it ensures the success of RNAi expression in vitro. Thus, our method can make an important contribution to disease-resistant RNAi studies.
CONCLUSION
In conclusion, we introduced a multi-resistance strategy against two major harmful diseases in pigs, FMDV and PRRSV. We expect that animals can be produced successfully according to previous studies. An in vitro shRNA verification method was suggested as a confirmation procedure before animal challenge testing. However, the in vitro method and in vivo test must be compared to confirm the effectiveness of the shRNAs. Further studies including our overall strategy and performed from a variety of perspectives are needed to identify resistance mechanisms against other critical diseases.
ACKNOWLEDGMENTS
This study was supported by the Next-generation BioGreen 21 Program (PJ0113002017), Rural Development Administration, Republic of Korea.
Notes
CONFLICT OF INTEREST
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.