Characterization and Profiling of Liver microRNAs by RNA-sequencing in Cattle Divergently Selected for Residual Feed Intake
Article information
Abstract
MicroRNAs (miRNAs) are short non-coding RNAs that post-transcriptionally regulate expression of mRNAs in many biological pathways. Liver plays an important role in the feed efficiency of animals and high and low efficient cattle demonstrated different gene expression profiles by microarray. Here we report comprehensive miRNAs profiles by next-gen deep sequencing in Angus cattle divergently selected for residual feed intake (RFI) and identify miRNAs related to feed efficiency in beef cattle. Two microRNA libraries were constructed from pooled RNA extracted from livers of low and high RFI cattle, and sequenced by Illumina genome analyser. In total, 23,628,103 high quality short sequence reads were obtained and more than half of these reads were matched to the bovine genome (UMD 3.1). We identified 305 known bovine miRNAs. Bta-miR-143, bta-miR-30, bta-miR-122, bta-miR-378, and bta-let-7 were the top five most abundant miRNAs families expressed in liver, representing more than 63% of expressed miRNAs. We also identified 52 homologous miRNAs and 10 novel putative bovine-specific miRNAs, based on precursor sequence and the secondary structure and utilizing the miRBase (v. 21). We compared the miRNAs profile between high and low RFI animals and ranked the most differentially expressed bovine known miRNAs. Bovine miR-143 was the most abundant miRNA in the bovine liver and comprised 20% of total expressed mapped miRNAs. The most highly expressed miRNA in liver of mice and humans, miR-122, was the third most abundant in our cattle liver samples. We also identified 10 putative novel bovine-specific miRNA candidates. Differentially expressed miRNAs between high and low RFI cattle were identified with 18 miRNAs being up-regulated and 7 other miRNAs down-regulated in low RFI cattle. Our study has identified comprehensive miRNAs expressed in bovine liver. Some of the expressed miRNAs are novel in cattle. The differentially expressed miRNAs between high and low RFI give some insights into liver miRNAs regulating physiological pathways underlying variation in this measure of feed efficiency in bovines.
INTRODUCTION
MicroRNAs (miRNAs) are small (~22 nucleotides) non-coding RNA that regulate gene expression by target messenger RNA (mRNA) in a sequence-specific manner, leading to either translational repression or degradation of targeted transcript. In animals, miRNAs target the 3′untranslated regions of mRNA by an RNA-induced silencing complex (RISC), and subject to the accuracy of the sequence complementarities, either repression of translation or cleavage of the mRNA target is achieved (Yu et al., 2007). MicroRNAs are now known to repress thousands of target genes and regulate cellular processes, including cellular proliferation, differentiation and apoptosis. The aberrant expression or alteration of miRNAs also contributes to a range of human pathologies, including diabetes and cancer (Lu et al., 2005).
Feed efficiency is an economically important trait in beef production and can be measured as residual feed intake (RFI). This is the difference between an animal’s actual feed intake recorded over a test period and its expected feed intake based on its size and growth rate (Koch et al., 1963). RFI takes into consideration feed required for daily weight gain of the animal as well as for maintenance of its metabolic weight; therefore, understanding of the molecular mechanism regulated RFI will help breeding sustainable and profitable animals in agriculture. Genome wide association studies have been carried out to identify gene markers associated with RFI in beef cattle (Sherman et al., 2009) and more than a hundred single nucleotide polymorphic (SNP) markers have been found associated with variation in RFI in beef cattle. However, a large proportion of SNPs markers are not located in annotated genes in bovine genome. Some of the most significant SNPs for RFI were in miRNAs motifs which suggest that these miRNAs could play an important role in regulation of RFI. Gene expression studies in cattle from high and low RFI divergent selection lines identified more than 160 differentially expressed genes (Chen et al., 2011). The variations of gene expression between high and low RFI cattle ranged from −1.5 to 0.8 fold change, and support the view that the phenotypic differences in RFI may due to level of gene expression instead of genes being switched on or off.
Profiling studies characterizing miRNAs encoded in livestock genomes in the last decade have found a wide and diverse range of conserved and species-specific miRNAs (Liu et al., 2009). In cattle, some initial characterizations of miRNAs have been carried out for various tissues including adipose tissue, mammary tissues (Gu et al., 2007), immune and embryonic tissues, pooled tissue, alveolar macrophages, ovarian, oocyte and testicular tissues (Miles et al., 2012), liver of dairy cows in the early postpartum period (Fatima et al., 2014). Liver is a central controller of metabolism and a major driver of whole animal oxygen consumption in mammals. However, little is known about miRNAs role in regulating key cellular and physiological pathways that may regulate RFI. In the current study, we profiled miRNAs abundance in liver tissue of Angus bulls from high and low RFI-selection lines by next-generation deep sequencing technology. We report liver miRNA-seq profiling study in beef cattle and their known and putative novel bovine miRNAs. We also report the differentially expressed miRNAs between high and low RFI-selection line cattle.
MATERIALS AND METHODS
Animal and liver biopsy sampling
Young Angus bulls resulting from approximately three generations of divergent selection for RFI were used in this study. The selection lines were established in 1993 at the Agricultural Research Centre, Trangie, NSW, Australia (Arthur et al., 2001; Donoghue et al., 2011). The same animals were used for the microarray experiment reported by Chen et al. (2011), and were approved by the University of New England Animal Ethics Committee, certificate no. (AEC 06/123) and followed the University of New England Code of Conduct for Research in meeting the Australian Code of Practice for the Care and Use of Animals. In brief, bulls were born in 2005 and, when approximately one year-of-age, their growth and feed intake were measured. Post-weaning RFI for each animal in the test group was calculated using a linear regression of daily feed intake on mean metabolic mid-test weight and average daily gain (Arthur et al., 2001). Based on the RFI test results, liver biopsies were taken (7 days after the end of the RFI test) from 30 animals with the lowest RFI and 30 animals with the highest RFI (Chen et al., 2011). On average, cattle from the low RFI line consumed about 2 kg less feed per day than cattle from the high RFI line (p<0.001; as shown in Table 1). Total RNA from liver was isolated using TRI reagent (Ambion, Applied Biosystems, Foster City, CA, USA) following the manufacturer’s protocol. The concentration and purity of the extracted RNA was checked by NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE, USA). The quality and integrity of RNA was further assessed with the RNA 6000 Nano Lab chip Kit using the Agilent 2100 Bioanalyzer (Agilent Technologies, USA). All RNA samples had a RNA integrity number (RIN) of 9.8 to 10) and were stored at −80°C until further analysis.
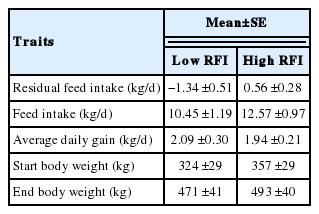
Feed efficiency performance of young Angus bulls selected for and against residual feed intake (RFI)
Small RNA library construction and sequencing
For small RNA library construction and deep sequencing, two pools of total RNA were constructed from 13 top high-RFI animals and 13 bottom low-RFI animals with equal quantities (1 μg of RNA) from each animal. Libraries of small RNAs were prepared using the “Preparing Samples for Small RNA Sequencing Using the Alternative v1.5 Protocol” supplied by the manufacturer and using 10 μg total RNA. The libraries were sequenced on the Illumina Genome Analyzer IIx using Single-Read Cluster Generation Kit v2 (Cat. no. FC-103–2001; Illumina) and 36 Cycle Sequencing Kit v4 (Cat. no. FC-104–4002). All sequencing data sets supporting the results in this study have been deposited in the publicly available NCBI’s Gene Expression Omnibus Database (http://www.ncbi.nlm.nih.gov/geo/). The data are accessible through GEO Series accession number GSE63691 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE63691).
Analysis of small RNA sequencing data
The raw sequence reads were processed with a Perl script to remove low quality reads and adaptor sequences and to count the reads of the Genome Analyzer (Illumina Inc., San Diego, CA, USA) into read-count files (read sequences and counts tab separated). Then the sequencing data were input into miRanalyzer (Hackenberg et al., 2011) to analyse the small RNAs. In brief, known bovine microRNAs were identified by mapping all sequence reads to the known bovine miRNAs database (miRBase v. 21) (Kozomara and Griffiths-Jones, 2011). Those reads matched to known bovine miRNAs were grouped and removed from the dataset so they could not erroneously be predicted as new miRNAs. Then, sequences reads mapped to non-bovine known miRNAs to identify homologous miRNAs for all other species in the miRBase. Genome sequences around the position of those mapped reads were extracted and the energetically best hairpin structures were retained as putative pre-miRNAs if they had: i) at least 19 base pairings in the secondary structure; and ii) at least 11 base pairings located in the read cluster region (number of pairings between putative mature and mature-star). Mapped reads were grouped as novel bovine homologue miRNAs and removed from data set. The remaining reads were further successively aligned to known transcriptome libraries such as RFam 10.1, GtRNAdb, RNAdb, and RefSeq (Gardner et al., 2009). To identify bovine-specific novel miRNAs, the remaining sequence reads were mapped to Bos taurus genome (BosTau6, UMD_3.1) using Bowtie (Langmead et al., 2009). The mapped reads were first clustered into putative mature miRNAs and pre-miRNAs. Putative candidate miRNAs reported based on at least three out of five models from the five different Random Forest models (Hackenberg et al., 2011). The secondary structure of pre-miRNAs was determined by RNAfold using minimum free energy (MFE) algorithm.
Differential expression analysis of miRNAs
The raw read counts of known bovine miRNAs and homologous miRNAs were imported as a count table into the Bioconductor DESeq package (Anders and Huber, 2010), which uses the negative binomial distribution model to test for differential expression in deep sequencing datasets. The normalization and variance estimation were based on the model of two conditions without replicate.
Prediction of putative miRNA targets
Potential gene targets for known miRNAs that differentially expressed between high and low RFI were identified using miRmap (Vejnar and Zdobnov, 2012). Only miRNAs target genes that highly scores (>80%) were selected for investigation as potential target genes. Also, all those genes expressed differentially between high and low RFI-selection line cattle which have been reported by Chen et al. (2011) were investigated for their potential miRNAs targets in the 3′UTR region.
RESULTS AND DISCUSSION
Profiling bovine miRNAs
In order to profile and identify novel and differentially-expressed miRNAs in the bovine liver of low and high RFI cattle, two small RNA libraries were constructed by Solexa sequencing. After trimming the adaptor sequence and filtering low-quality sequences, a total of 23,628,109 sequences of short reads were obtained for low and high RFI libraries. As presented in Table 2, there were 10,820,087 sequences reads for the low RFI library (high feed efficiency cattle) and 12,808,022 sequence reads for the high RFI (low feed efficiency cattle). All sequence reads were aligned against the bovine miRNAs database (miRBase v.21). Importantly, high proportion 74.5% and 83.7% of sequence reads of the two RFI libraries have aligned to the bovine genome (UMD 3.1). Over half of each library reads (56% to 57%) have showed an exact match to the known bovine mature miRNAs, as shown in Table 2. These results demonstrated the high quality of our small RNAs sequencing read libraries. However, the presence of other small RNAs, such as ribosomal fragments, tRNA, snRNA, and mtRNA was approximately 12% and 17% of mapped sequence reads of low and high RFI libraries respectively. Sequence reads that matched to homologous miRNAs of other species reported in the miRBase constituted 1% of mapped reads (Figure 1).
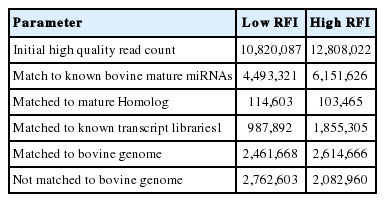
Summary of miRNA sequences present in low and high RFI libraries
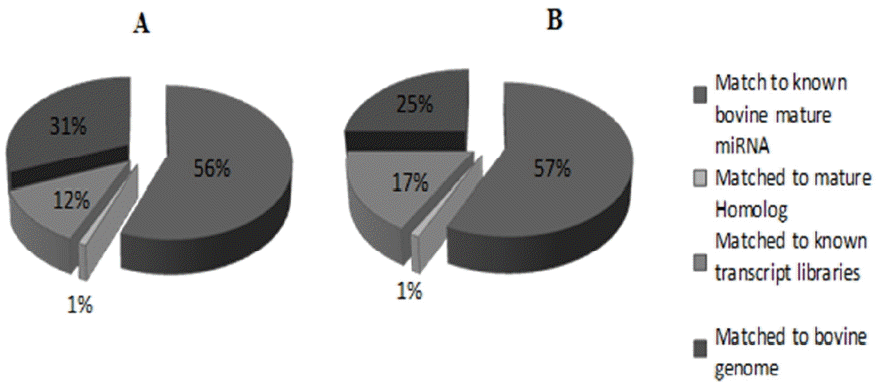
Distribution of high quality reads that mapped to the bovine genome in the low RFI line (A) and high RFI line (B) cattle. RFI, residual feed intake.
Unique known mature bovine miRNAs
In total 305 known mature miRNAs detected in two libraries and the full profile of unique bovine mature miRNAs expressed in liver tissue was listed in Additional files, Supplementary Table S1. Table 3 lists only the most abundant miRNAs (>100 read count per million mapped read [RCPM]) from 305 known mature miRNAs. About 40% (305 out of 807) of known bovine mature miRNAs were expressed in the liver tissue. The proportion of expressed miRNAs in bovine liver is similar to the level of expressed miRNAs in human and mouse liver (Kornfeld et al., 2013). In contrast, primary bovine mammary epithelial cells expressed only 20% of known bovine miRNA (Lawless et al., 2013).
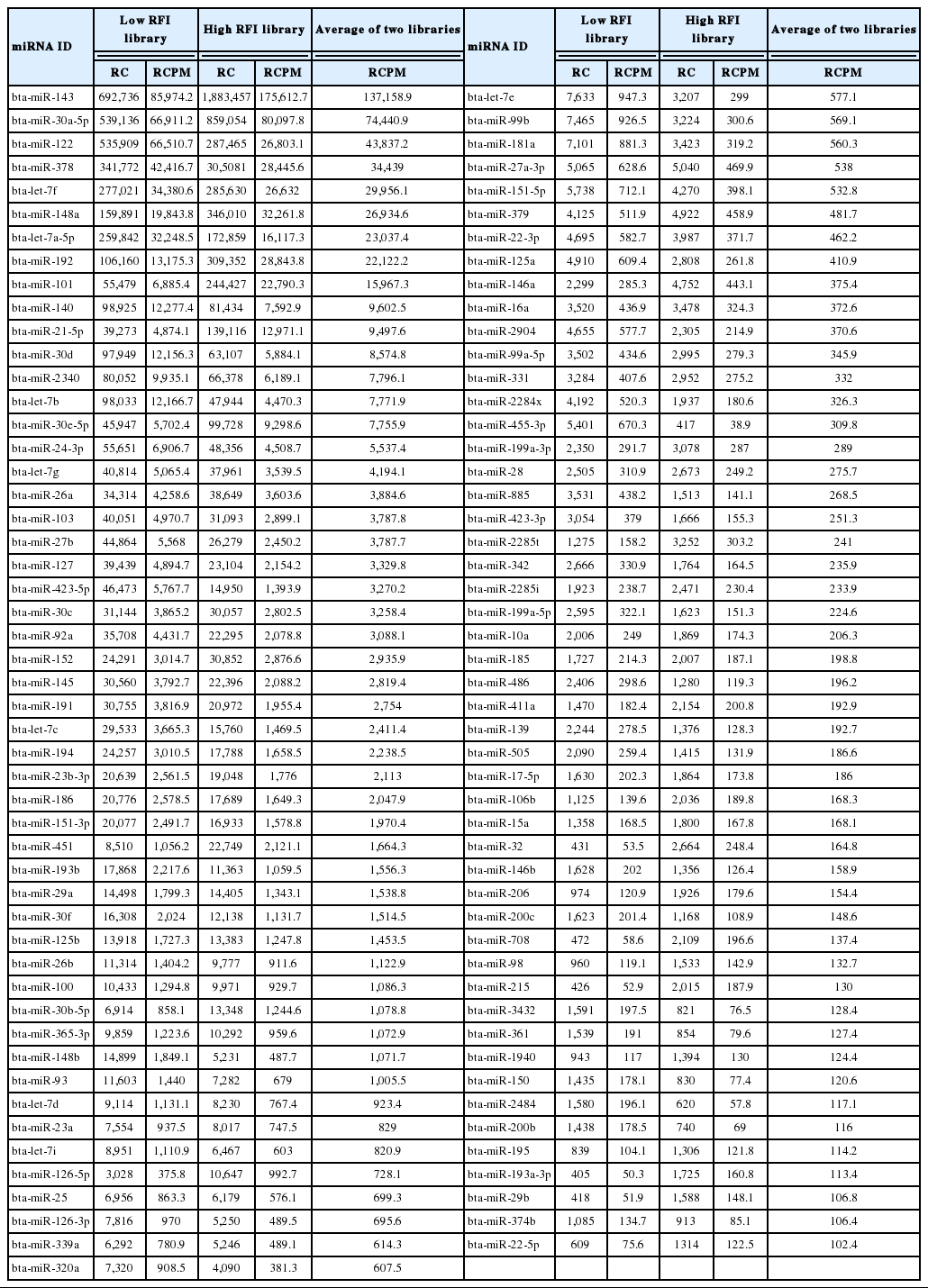
The most abundant bovine mature miRNAs in the high and low RFI sequence libraries from liver tissue
A very broad range in expression levels of miRNAs was found ranged from 137,000 RCPM to less than one RCPM (Additional files, Supplementary Table S1). Only a small number of miRNAs were expressed at very high levels, such as bta-miR-143, bta-miR-30a, and bta-miR-122, and the majority of miRNAs being expressed at medium and low levels. However, the top 10 most abundantly expressed miRNAs families (bta-miR-143, bta-miR-30, bta-miR-122, bta-miR-378, bta-let-7, bta-miR-148, bta-miR-192, bta-miR-101, bta-miR-140, and bta-miR-21) constituted approximately 78.4% of the total mapped mature known bovine sequence reads. These findings are in agreement with studies of miRNAs profiling which revealed that small number of abundant miRNAs make up a high portion of the total miRNAs pool (Tripurani et al., 2010; Fatima et al., 2014).
Among the highly expressed known miRNAs detected in the bovine liver in our study, miR-143 was the most abundant miRNA, constituting approximately 25% of the total expressed miRNAs. In human and mouse liver the most abundant miRNA is miR-122 (Hu et al., 2012; Rottiers and Näär 2012), which makes up 70% of the total mouse liver miRNA population (Wen and Friedman, 2012). Our results showed that bovine bta-miR-122 was the third most expressed miRNA, comprising approximately 8% of the total known bovine miRNA population.
Previously, tissue-specific miRNAs were reported in dairy and beef cattle (Jin et al., 2009; Tripurani et al., 2010) using cloning and sequencing small-RNA libraries. One of those tissue-specific miRNAs was bta-miR-9. Bovine miR-9 was identified and reported as brain-specific miRNA (Jin et al., 2009). Similarly, bta-miR-1, bta-miR-133a, and bta-miR-206 have been reported as muscle-specific miRNAs in bovine (Jin et al., 2009). In the current study, these four miRNAs have expressed in bovine liver with sensible expression levels in the two high and low RFI libraries. The expression levels of bta-miR-1, bta-miR-206, and bta-miR-133a were 7,081, 1,450, and 689 RCPM respectively. These results suggest that previous reports on tissue specificity of these miRNAs might be limited by the tissues surveyed and/or sensitivity of the techniques that used.
Novel bovine miRNA
Novel isomiRs: riations with respect to the reference miRNA sequence (isomiRs) were named previously as mature-star (miRNA*), and were considered as “minor” products from pre-microRNA sequence. In miRNAs biogenesis, it was often inferred to be degraded and only the dominant arm (sense strand) to be incorporated into the RISC complex. However, there is strong evidence that these isomiRs are heterogeneous variants of canonical miRNAs species and are of functional importance (Yang et al., 2011). We adopted the new nomenclature using −5′p/−3′p for the name of isomiRs miRNA in the bovine library (Figure 2). Fifty novel isomiRs were detected in the present study, which have not been observed previously in the bovine miRNA database. Four isomiRs, bta-mir-143-3p, bta-mir-335-3p, bta-mir-136-3p, and bta-mir-2284w-3p have identified to have higher read counts than the corresponding known mature miRNA reported in the bovine miRBase, as illustrated in Table 4. For instance, bta-mir-143-3p was expressed by approximately 605 RCPM, while mature bta-miR-143-5p was expressed by only about 19.5 RCPM in the average of the two libraries. Therefore, more studies still needed to evaluate which isomiRs is dominant and functional to improve the bovine miRNA annotation.

Precursor miRNA structures, the 5′p (mature sequence) is highlighted in red and 3′p (miRNA* sequence) is highlighted in black bold fonts.
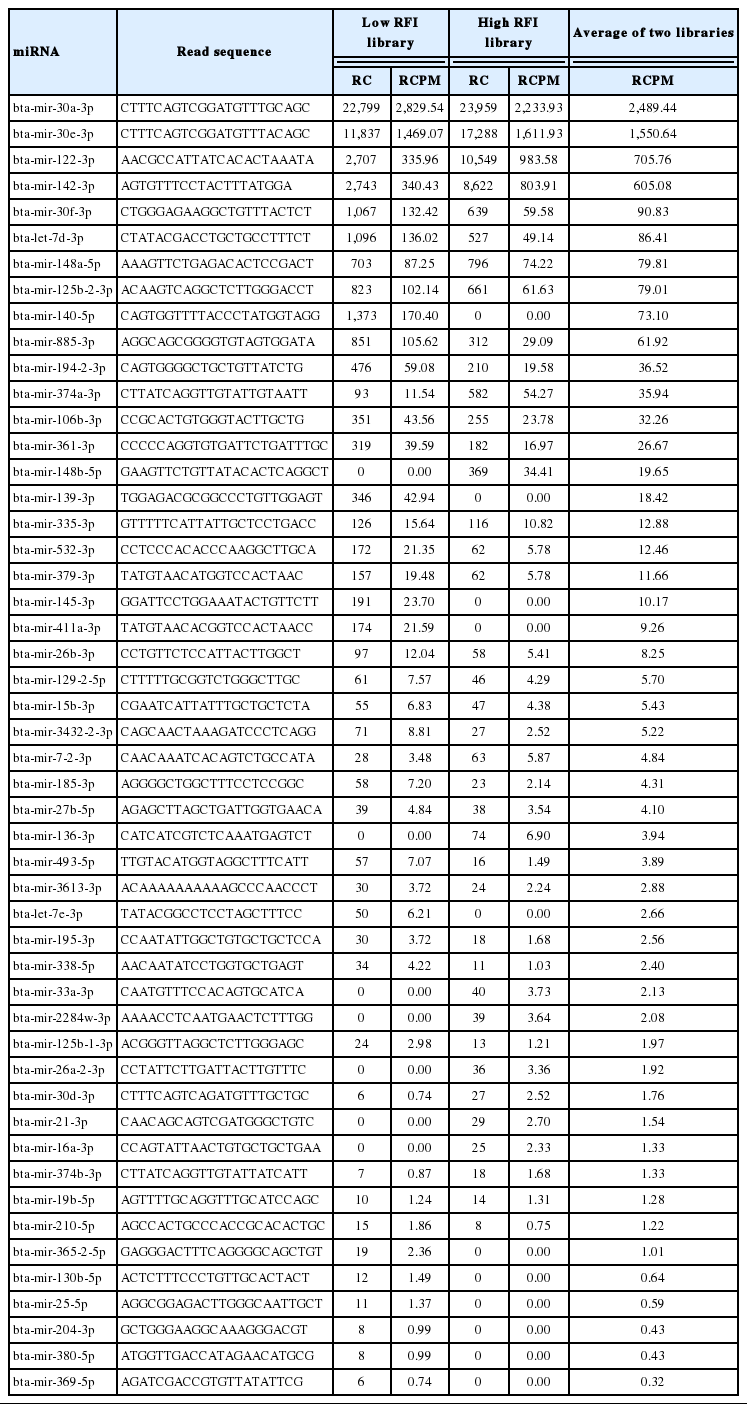
Novel isomiRs detected in low and high RFI libraries and their related expression (read counts)
Putative homologous bovine miRNAs
Bovine putative homologous miRNAs are those miRNAs that have been reported in other species but not in bovine miRNAs database yet. After removing all reads matched to known bovine miRNAs, the remaining reads were then aligned to non-bovine sets of known mature miRNAs from other species (miRBase v. 21). There were 61 detected putative homologous miRNAs which have distinct hairpin loci in the bovine genome (Table 5). These putative novel homologous miRNAs sequences and related genomic locations are presented in Additional files, Supplementary Table S2.
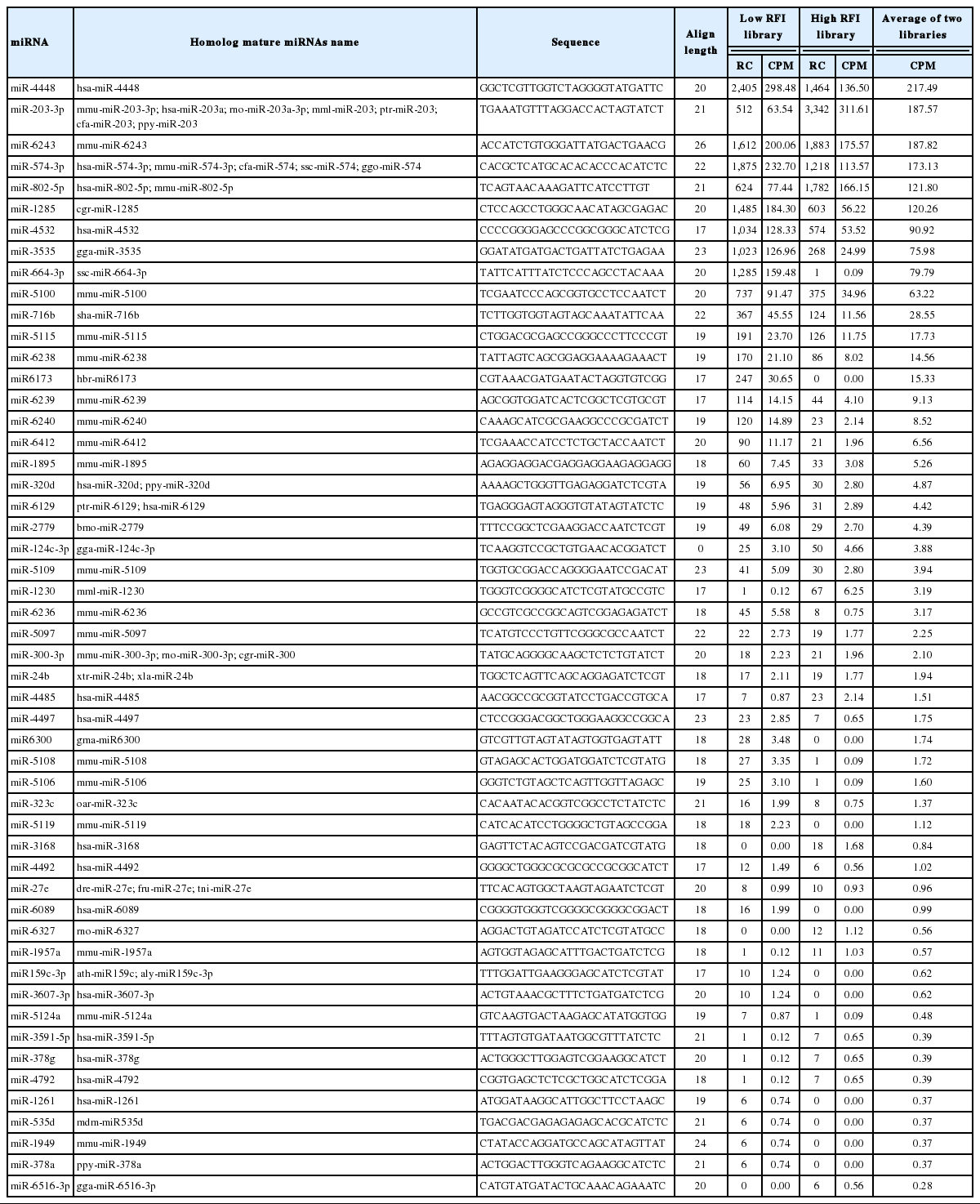
Unique putative homolog miRNAs identified in bovine liver tissue
Detection of putative novel bovine microRNAs
More than quarter of the reads (25% from high and 31% from low RFI libraries) that did not matched to known bovine miRNAs matched to the bovine genome. To identify novel miRNAs, we extracted the candidate pre-miRNA structure, based on the location of clusters of mature miRNA on the genome, to select the energetically best candidate if they were having: i) at least 19 base pairings in the secondary structure, and ii) at least 11 base pairings located in the read cluster region (the number of pairings between 5′p and 3′p). Then five different models (Hackenberg et al., 2011) were used to predict whether a given candidate was likely to be a miRNA. We considered a candidate as a novel miRNA only if at least three out of five models were positive. Twelve putative new miRNAs were discovered in the present study. After realigning these putative novel consensus sequences to known mature miRNAs in other species, two candidates (miR-664-3p and miR-802-5p) were found to be homologous to other species. Ten novel bovine-specific candidate miRNAs with their sequence, genomic location, and the number of the models that predicted each novel miRNA candidate are presented in Table 6. These ten novel bovine miRNAs represented by 5,437 and 1,097 reads in the low RFI and high RFI libraries, respectively. The sequences and secondary structure of those 10 novel bovine-specific miRNAs candidates and related pre-miRNAs with the MFE, which have been predicted by RNAfold program, are illustrated in Additional files, Supplementary Figure S1. Novel candidate 1 and 2 have identical mature sequences but resulted from distinct precursor sequences located in different genomic region.
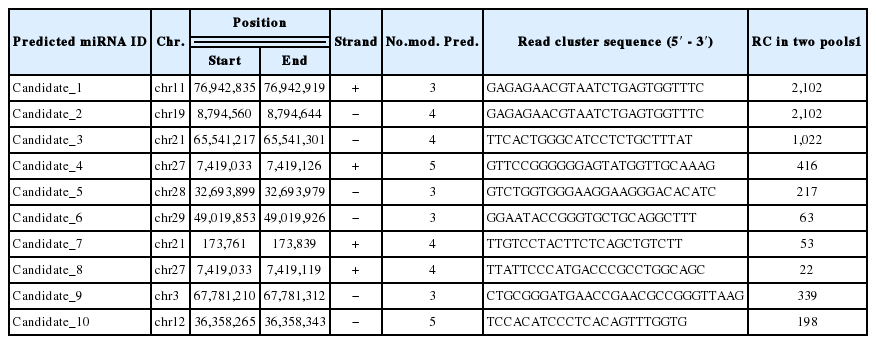
Novel bovine miRNAs detected in the bovine liver tissue, with sequence, genomic location and their related expression (read counts)
Differentially expressed miRNA between high and low RFI libraries and their target genes
To understand the role of miRNA in regulation of residual intake, we first identified differentially expressed miRNAs from high and low RFI selection lines. As miRNAs play important roles in the regulation of almost every biological process in eukaryotes; these differentially expressed miRNAs could play important roles in regulation of the physiological processes and pathways involved in variation in RFI in beef cattle. To define the differentially expressed miRNAs, we consider only those miRNAs with high to modest expression with at least 100 RCPM and folds changes>2. This approach was adapted in earlier microarray experiment and was less stringent than using p<0.05 due to lack of replicates. A total of 49 unique miRNAs were identified to be differentially expressed between the high RFI line and low RFI line cattle (Table 7). More than two thirds (33 out of 49) of differentially expressed miRNAs were up-regulated in high RFI animals. Specifically, six miRNA (miR-203-3p, bta-miR-32, bta-miR-215, bta-miR-708, and bta-miR-101) that reached p<0.05 were all up-regulated in high RFI animals.

miRNAs differentially expressed between the high and low RFI libraries
Importantly, many up-regulated miRNAs in high RFI animals played important roles in metabolic homeostasis including glucose and lipid metabolism. These overexpression patterns were similar to the patterns observed in obesity mouse models and human subject. For example bta-miR-143, the most expressed miRNA in bovine liver, was up-regulated in high RFI cattle. It has been reported that hepatic miR-143 were up-regulated in obesity mouse models whether they are genetic or dietary induced. The overexpression of hepatic miR-143 impaired insulin-stimulated AKT activation and glucose homeostasis by targeting insulin signalling and its regulation (Jordan et al., 2011). Bta-mir-122-3p was highly expressed in bovine liver and is up-regulated in high RFI cattle. MiR-122 was the first miRNA to be linked to metabolic control and affect hepatic cholesterol, lipid metabolism and the maintenance of liver cell differentiation (Lewis and Jopling, 2010).
A bovine homologous of miR-802-5p was identified in our study and was up-regulated in high RFI cattle. MiR-802 has been reported up-regulated in the liver of two obese mouse models and obese human subjects (Kornfeld et al., 2013). Overexpression of miR-802 in mice causes impaired glucose tolerance and attenuates insulin sensitivity by suppressing its target gene HNF1 Homeobox B (Hnf1b).
Bta-miR-29b was up-regulated in high RFI. The function of miR-29 has been reported to regulate glucose transport in liver, muscle and adipose (Pandey et al., 2011). Bta-miR-19b, bta-miR-101, bta-miR-106b, and bta-miR-142-3p were up-regulated in high RFI cattle. Recently studies showed their expression in steers liver were highly influenced by energy density of the diet (mainly lipid levels in the diet) (Romao et al., 2012).
To understand miRNAs expression and their target genes, we searched all the potential miRNAs target sites in the 3′UTR region of 161 differentially expressed genes identified by Chen et al. (2011) by using miRmap (Vejnar and Zdobnov, 2012). Then we examined if these potential miRNAs were expressed differentially in liver between high and low RFI cattle.
We found 36 differentially-expressed genes in liver (Chen et al., 2011) containing putative miRNAs target sites (Table 8). Many genes have multiple miRNAs target sites such as helicase with zinc finger (HELZ), espin (ESPN), cytochrome P450 family 2 subfamily C member 18 (CYP2C18), snail family transcriptional repressor 2 (SNAI2), and superoxide dismutase 3 (SOD3). Five down-regulated miRNAs (bta-miR-424-5P, bta-miR-19b, bta-miR-29b, bta-miR-30b, and bta-miR-2285) having sites binding to the mRNA of 12 distinct genes calponin 1 (CNN1), atypical chemokine receptor 3 (CXCR7), endothelin receptor type (BEDNRB), fibrinogen alpha chain (FGA), insulin like growth factor binding protein 3 (IGFBP3), regulator of G-protein signaling 2 (RGS2), periostin, osteoblast specific factor (POSTN), monoamine oxidase (AMAOA), collagen type IV alpha 6 (COL4A6), dehydrogenase/reductase (SDR family) member 3 (DHRS3), solute carrier family 22 (organic anion transporter), member 7 (SLC22A7). These genes were up-regulated in low RFI animals (Chen et al., 2011). Four genes, BEDNRB, IGFBP3, POSTN, and DHRS3 were up-regulated in low-RFI cattle (Chen et al., 2011) and have bta-miR-19b putative target sites in their 3′UTR region. Take together we believe these differentially expressed genes play important roles regulating RFI in beef cattle.
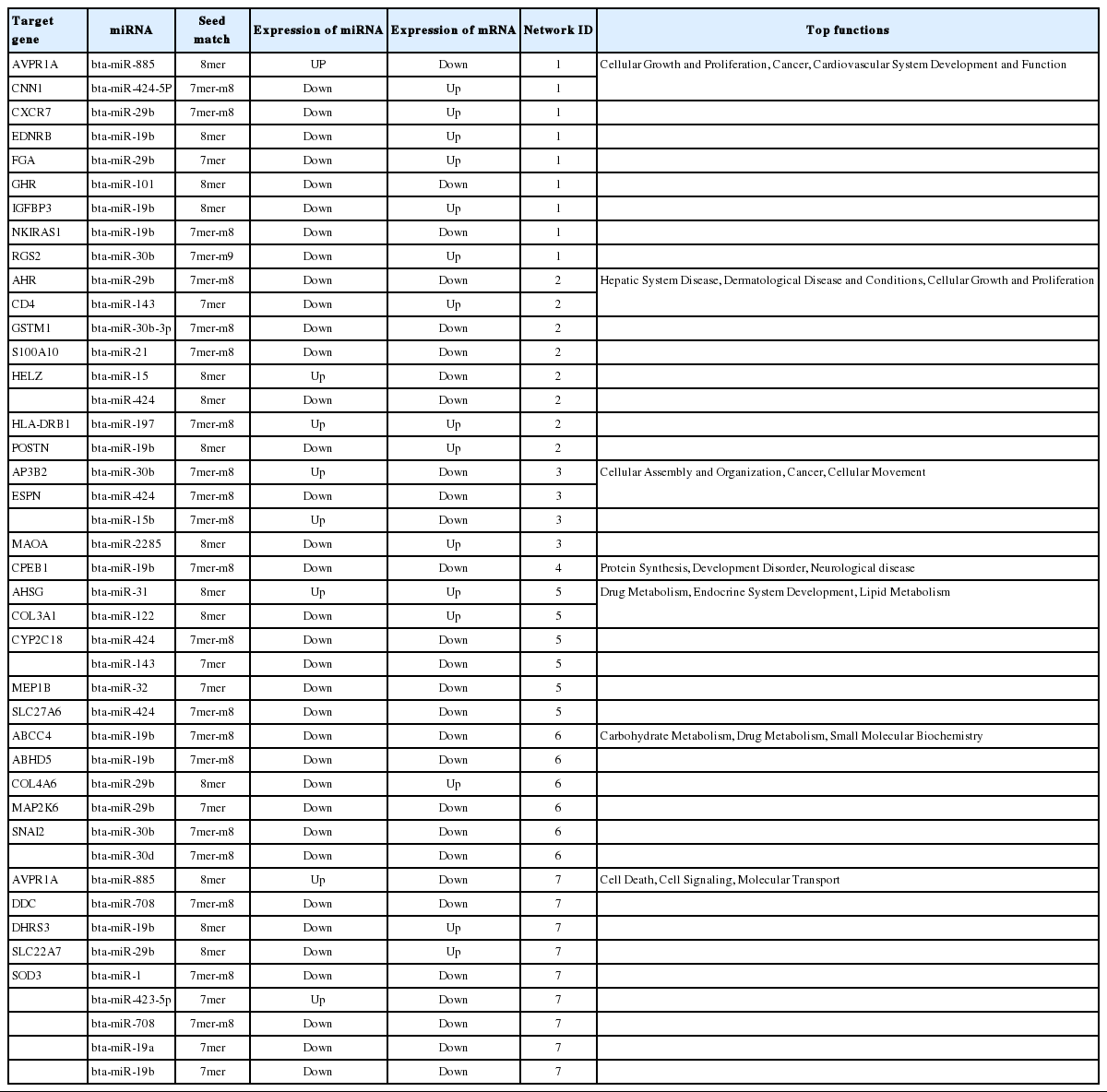
However, we acknowledge the differentially expressed genes detected in this study are based on pools with limited statistic power. Follow up studies are need for validating the differentially expressed miRNAs and their function.
In conclusion, RFI as a measure of feed efficiency is influenced by several physiological systems including basal metabolic rate, energy balance, the regulation of growth and development, regulation of feed intake and homeostatic control of body mass. Our study revealed a comprehensive miRNA population in bovine liver. We identified 305 known bovine miRNAs, 50 novel isomiRs, 52 homologous miRNAs, and 10 novel miRNAs. We further revealed that many up-regulated miRNAs in high RFI cattle showed a similar expression pattern as found in a mouse obesity model and have functions related to glucose and lipid metabolism. We demonstrated the expression of miRNAs have effects on their target genes expression. Combining the patterns of miRNA and mRNA expression will provide further power to understanding the molecular mechanisms that regulate feed efficiency in beef cattle.
Supplementary Information
ACKNOWLEDGMENTS
This work was funded by the former Cooperative Research Centre for Beef Genetic Technologies and the University of New England. Wijdan Al-Husseini was supported by the University of Babylon/Ministry of Higher Education and Scientific Research (MOHESR)/Iraq.
Notes
CONFLICT OF INTEREST
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.