




INTRODUCTION
Although whole genome sequences, including those of avian species, are publically available and massive information can be extracted using bioinformatics tools, knowing the structure of a predicted gene and its function(s) does not provide a comprehensive understanding of the biological activity and regulatory mechanism of all of the predicted target genes in a cell or living organism. Thus, in vivo and in vitro target validation approaches are necessary to accurately and precisely evaluate the biofunctional activity of predicted target genes.
Technical platforms for functional genomics analysis are mainly divided into gain-of-function and loss-of-function strategies. As a gain-of-function strategy, foreign gene delivery and expression can be applied using the various transgene expression promoters and vector systems that have been developed in mammals and birds [1ŌĆō4]. The simian virus 40, cytomegalovirus (CMV), and CMV early enhancer/chicken ╬▓-actin (CAG) promoters are frequently and widely used for constitutive and strong transgene expression [1,3,4]. The CMV promoter, which is derived from the human CMV immediate early enhancer and promoter, is commonly used for specific gene functional analyses in vitro and in vivo [1,4]. The CAG promoter is a synthetic promoter consisting of the CMV early enhancer element/promoter and the first exon and first intron of the CAG gene with the splice acceptor of the rabbit ╬▓-globin gene [3,5,6]. Additionally, the non-virus-originated human elongation factor 1 alpha promoter was developed for efficient high-level transgene expression [2]. Another versatile approach is an inducible transgene expression system. To date, various inducible expression systems for transgenes have been developed involving complicated inducers such as metal ions, steroid hormones, and tetracycline [7ŌĆō9]. The tetracycline resistance operon from Escherichia coli can efficiently control transgene expression in mammals [9]. An alternative cumate-regulated inducible system shows dynamic transgene expression inducibility that can be tightly modulated and is reversible in the absence of cumate [10]. Since transgene repression and expression can be efficiently regulated by the absence or presence of a non-toxic cumate molecule, this induction system can be widely applied to functional genomic analyses in poultry.
To eliminate a specific target gene, the clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9-mediated genome editing technical platform, a powerful and versatile technology, was recently developed and has been frequently and widely adapted for functional genomics studies because of its simple and efficient genome-programmable advantages [11,12]. To achieve site-specific DNA recognition and cleavage, the Cas9 enzyme first complexes with a guide RNA (gRNA), which is complementary to the targeted sequences. The nuclease domain in Cas9 then cuts the targeted DNA strands and generates a double-stranded break [11,12]. Finally, a nucleotide deletion or insertion is produced during non-homologous end joining, creating a frameshift mutation [11,12].
Rubin et al [16] identified two major genes correlated to chicken domestication process through selective-sweep analysis of chicken genome; thyroid stimulating hormone receptor (TSHR) and SH3 domain containing ring finger 2 (SH3RF2) gene. They found two deletions in the coding sequences suggesting that they are functionally important. Subsequently, these deletion mutations would be directly applicable to animal breeding and enhance the importance of the domestic chicken [13]. However, the biofunctional study for these candidate genes has not been conducted yet. In this study, we knocked out the SH3RF2 gene in quail myoblast cells using the CRISPR/Cas9 technique and investigated the functional activity of SH3RF2 during myotube differentiation.
MATERIALS AND METHODS
Quail myoblast cell culture and induction of myotube differentiation
Quail myoblast (QM7) cells (American Type Culture Collection, Manassas, VA, USA) were maintained at 37┬░C in an atmosphere of 5% CO2 and 60% to 70% relative humidity in Medium 199 containing 10% fetal bovine serum (FBS; HyClone, Logan, UT, USA), 2% chicken serum (Sigma-Aldrich, St. Louis, MO, USA), and 1├Ś antibiotic-antimycotic (Invitrogen, Carlsbad, CA, USA) by subculturing cells at 70% confluency. For the induction of myotube differentiation at 90% confluency, the differentiation medium containing 0.5% FBS and 1├Ś antibiotic-antimycotic was changed and half of the medium was replaced with fresh differentiation medium daily.
The CRISPR/Cas9-mediated knock out of SH3RF2 and fluorescence-activated cell sorting
To knock out SH3RF2, 7.5 ╬╝L of Lipofectamine 3000 Reagent was diluted in 250 ╬╝L of OPTI-MEM (Invitrogen, USA), and 2.5 ╬╝g each of the Cas9-green fluorescent protein (GFP) co-expression plasmids (Sigma-Aldrich, USA) and SH3RF2 gRNA were mixed with Lipofectamine P3000 Reagent in 250 ╬╝L of OPTI-MEM at room temperature. After incubation for 5 min, the two mixtures were combined and incubated for 20 min. The complex mixture was gently pipetted and dropped into a six-well plate containing QM7 cells at 70% to 80% confluency. After incubation at 37┬░C in 5% CO2 for 4 h, the cells were gently washed with phosphate-buffered saline (PBS) three times and fresh culture medium was added. One day after lipofection, GFP-expressing cells were sorted using a FACSAria III cell sorter (Becton, Dickinson and Co., Franklin Lakes, NJ, USA). Following harvest using 0.05% trypsin-ethylenediaminetetraacetic acid (Invitrogen, USA), the cells were resuspended in PBS containing 0.1% bovine serum albumin and strained through a 40 ╬╝m cell strainer for fluorescence-activated cell sorting (FACS; BD Falcon; Becton, Dickinson, and Co., Franklin Lakes, NJ, USA). After sorting, the cells were regrown in culture media for subsequent experiments. To isolate single cell-derived sublines, each well-isolated single colony was isolated and subcultured using smooth silicone grease and a cloning cylinder.
T7 endonuclease I assay and genotyping by T-vector cloning and sequencing
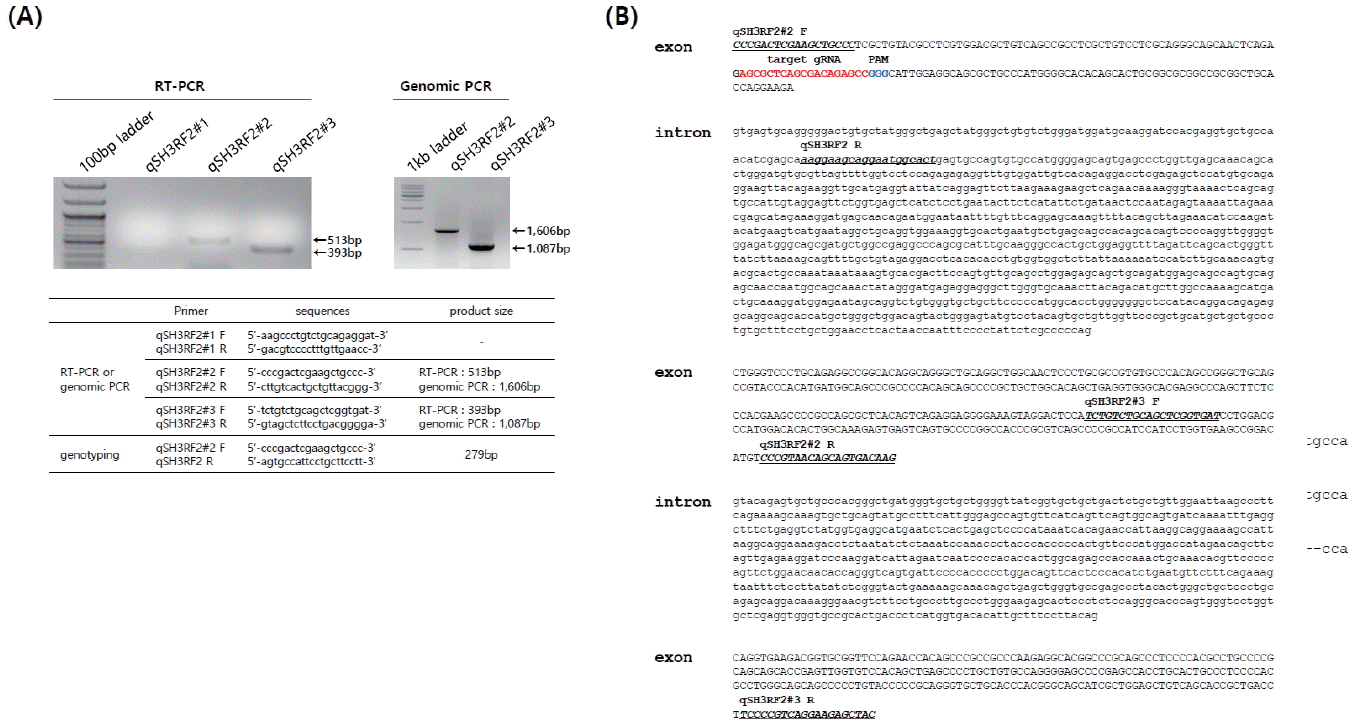
For the T7 endonuclease I (T7E1) assay, the genomic region of the targeted site was amplified with a specific primer set. The amplicons were re-annealed to form a heteroduplex DNA structure after denaturation. Subsequently, the heteroduplex amplicons were treated with 5 U of T7E1 (New England Biolabs, Ipswich, MA, USA) for 15 min at 37┬░C and then analyzed by agarose gel electrophoresis. Genomic polymerase chain reaction (PCR) was performed using an initial incubation at 94┬░C for 5 min, followed by cycles of denaturation, annealing, and extension using the SH3RF2 genotyping primer set (Figure 1A). The reaction was terminated with a final incubation at 72┬░C for 5 min. To confirm the target locus mutation, the PCR amplicons were cloned into pGEM-T easy (Promega, Madison, WI, USA) and sequenced using an ABI 3730XL DNA Analyzer (Applied Biosystems, Foster City, CA, USA).
De novo assembly of the RNA-Seq data for the quail transcripts
Gene expression levels were estimated through the Trinity pipeline (version 2.2.0) using RNA-Seq by expectation maximization (RSEM) as the transcript quantification method. The Trinity pipeline was employed for non-model organisms, mapping the reads into an assembled consensus without a genomic reference to measure transcriptome levels. Trinity allows for the identification of transcript isoforms in non-model species. Prior to assembly, we employed Trimmomatic (version 0.35) for the removal of adapter sequences. Next, we performed assembly and mapping as follows. To construct the reference consensus and assemble the transcriptome, we combined left and right reads in paired-end reads using each sample. Then, using combined left and right reads, we created the assembled transcriptome with ŌĆśtrinityrnaseq-2.2.0/TrinityŌĆÖ by applying the default settings. Next, we performed read mapping onto the assembled transcriptome (bowtie2) and abundance estimation (RSEM) using align_and_estimate_abundance.pl applying the ŌĆś--est_method RSEM --aln_method bowtie --trinity_modeŌĆÖ options. Gene annotation was performed using TransDecoder, which is implemented within Trinity, and several developer-recommended gene annotation tools such as blastx and blastp were applied. Finally, using Trinotate, we extracted information on annotated genes by applying the default settings. To define known transcriptomic regions, we used Trinotate (http://trinotate.github.io) applying the default settings.
Statistical analysis
Using the trimmed mean of M-values normalized expression as the response, the analysis of deviance model was employed to test for significance between the knockout (KO) and control groups, as follows:
Where i = {control, KO} with control samples serving as the baseline. Negative-binomial assumption was considered a response variable to solve the over-dispersion problem in count data. Under the null hypothesis (H0: Group = 0), a likelihood ratio test was performed and the p-values were adjusted based on the false discovery rate. Here, a 1% significance level was considered significant.
Reverse transcription-polymerase chain reaction and quantitative reverse transcription-PCR
Total RNA from undifferentiated regular QM7 (rQM7) and SH3RF2 KO QM7#4 cells was isolated using TRIzol Reagent (Invitrogen, USA) according to the manufacturerŌĆÖs instructions. RNA quality was examined by agarose gel electrophoresis and quantity was determined on a NanoDrop 2000 (Thermo Scientific, Delaware City, DE, USA). cDNA was synthesized from RNA using the Superscript III First-Strand Synthesis System (Invitrogen, USA). Each 20-╬╝L reverse transcription (RT)-PCR reaction contained 2 ╬╝L of cDNA, 2 ╬╝L of PCR buffer, 1.6 ╬╝L of a dNTP mixture (2.5 mM), 1 U of Taq DNA polymerase, and 10 pmol of the forward and reverse primers (Figure 1A). PCR was performed with an initial incubation at 94┬░C for 5 min, followed by 35 cycles of 94┬░C for 30 s, 60┬░C for 30 s, and 72┬░C for 30 s. The reaction was terminated with a final incubation at 72┬░C for 5 min and the products were analyzed by agarose gel electrophoresis. For the quantification of paired box 7 (Pax7) transcripts, quantitative (q)RT-PCR was performed using the iCycler iQ real-time PCR Detection System (Bio-Rad, Hercules, CA, USA) and Eva Green (Biotium, Fremont, CA, USA). The conditions were as follows: 94┬░C for 5 min, followed by 35 cycles of 94┬░C for 30 s, 60┬░C for 30 s, and 72┬░C for 30 s. Melting curve profiles for the amplicons were analyzed. Quantitative real-time PCR data for the target genes were normalized relative to ╬▓-actin expression and were calculated using the 2ŌłÆ╬ö╬öCt method.
Western blot analysis
Total protein was extracted with 1├Ś radioimmunoprecipitation lysis buffer and separated on a 10% polyacrylamide gel followed by transfer to a nitrocellulose membrane (Bio-Rad, USA). The primary antibodies used were mouse anti-╬▓-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and anti-MyoD (Santa Cruz Biotechnology, USA). Horseradish peroxidase-conjugated anti-mouse IgG (anti-╬▓-actin) or anti-rabbit IgG (anti-MyoD antibody) was used as a secondary antibody. The blots were treated with enhanced chemiluminescence substrate solutions and exposed using a ChemiDoc XRS System (Bio-Rad, USA).
RESULTS AND DISCUSSION
Identification of the quail SH3RF2 gene structure
Although quail is a major livestock in the poultry industry, there are no available genome sequences in the public domain, although the draft quail genome was published in 2013 [14]. Thus, we first cloned and identified the sequences and structure of quail SH3RF2. To identify the transcript and genomic structure of quail SH3RF2, we designed primer sets based on chicken SH3RF2 and amplified the target sites by RT-PCR and genomic PCR (Figure 1A). The primer sets, except for qSH3RF2#1, produced products by both RT-PCR and genomic PCR. These products were then sequenced and analyzed after T-vector cloning. From a sequence analysis of the PCR products, we partially arranged three exons and two introns (Figure 1B). SH3RF proteins consist of multi-scaffold domains as well as a really interesting new gene domain [13,15]. SH3RF family members (three types: SH3RF1ŌĆō3) are involved in cell survival and apoptosis [15,16]. Wilhelm et al [15] reported that SH3RF2 acts as an anti-apoptotic regulator of the c-Jun N-terminal kinase pathway. Additionally, SH3RF2 was reported as an oncogene product that is overexpressed in human cancers by regulating p21-activated kinase 4 protein stability [13]. In chicken, Rubin et al [16] reported a deletion in the first exon of the orthologous chicken gene, found particularly in high growth lines, suggesting that a loss-of-function of SH3RF2 is strongly related to growth performance and body weight. Under the selection process during chicken domestication, this deletion mutation has been fixed in high growth selection lines [13]. Because SH3RF2 is expressed in brain and muscle [13], we knocked out SH3RF2 in quail myoblast cells to investigate its functional activity during myotube differentiation.
Design of a gRNA for the target site and construction of the Cas9-GFP and gRNA expression vectors
Based on the sequence information, the target gRNA site was designed in the exon region of SH3RF2 with a protospacer adjacent motif sequence (Figure 1B). To express GFP-fused Cas9 by 2A sequences and gRNA, an all-in-one expression vector was constructed (Figure 2A). The expression of Cas9-GFP and gRNA was controlled by the CMV and U6 promoters, respectively (Figure 2A). U6 promoter-gRNA was inserted into the Cas9-GFP expression vector by digestion with MluI and ligation. The orientation of the gRNA construct was confirmed by sequencing. The overall experimental scheme is presented in Figure 2B.
CRISPR/Cas9-mediated SH3RF2 KO and establishment of single cell-derived sublines
After transfection of the Cas9-SH3RF2 gRNA expression vector into QM7 cells, GFP-positive cells were identified by FACS and a single cell was isolated followed by in vitro expansion. To examine the induced mutations in the single cell-derived sublines, a T7E1 assay was conducted and three sublines showed T7E1 cleavage (Figure 3A). To identify the mutant genotypes, the targeted locus was amplified and sequenced (Figure 3B). SH3RF2 KO QM7#4 had the mutant genotype, containing 61 and 155 nucleotide deletions, whereas SH3RF2 KO QM7#1 showed a mixed genotype, containing 15, 33, and 70 nucleotide deletions (Figure 3B). Thus, SH3RF2 KO QM7#4 was used in subsequent experiments to examine myotube differentiation.
Differentiation and characterization of SH3RF2 KO QM7#4
Regular QM7 (rQM7) and SH3RF2 KO QM7#4 cells had a similar morphology in the undifferentiated state (Figure 4A). Additionally, there was no significant difference in the expression of Pax7, which is a critical marker of undifferentiated myoblasts (Figure 2B). These results indicated no significant phenotypic difference in SH3RF2 KO QM7 cells compared to rQM7 cells. To confirm myogenic gene expression during the differentiation process, Western blotting was performed after 6 days of differentiation. The expression of MyoD was examined in both rQM7 and SH3RF2 KO QM7#4 cells on different days of differentiation (Figure 4B). In addition, under differentiation conditions, both rQM7 and SH3RF2 KO QM7#4 cells underwent differentiation and constantly formed myotubes (Figure 4C). Similar to the differentiated rQM7 cells, the differentiated SH3RF2 KO QM7#4 cells clearly showed nuclear fusion and the presence of multiple nuclei in the induced myotubes (Figure 4C). There was no significant difference between the percentage of the relatively differentiated area in rQM7 and SH3RF2 KO QM7#4 cells (Figure 4D).
Global gene expression analysis by RNA sequencing in SH3RF2 KO QM7#4 cells
Figure 5 shows the reciprocal interactions of differentially expressed genes in undifferentiated rQM7 and MyoD KO#4 cells. In the undifferentiated stage, SH3RF2 mainly interacts with structure specific recognition protein 1 (SSRP1), and, subsequently, SSRP1 regulates many downstream cascade genes (Figure 5A). SSRP1 is a subunit of the faciliates chromatin transcription complex; it forms a heterodimeric complex with SPT16 to regulate transcriptional elongation and possibly modulate DNA replication [17ŌĆō19]. Since replication defects have been observed in SSRP1 mutants, a role for SSRP1 in DNA repair by homologous recombination was also demonstrated in mammalian cells [20]. Additionally, SSRP1 facilitates microtubule growth and bundling for mitosis [21]. The knockdown of SSRP1 inhibits the growth of microtubules, causing the disorganization of spindle structures and disruption of chromosome movement, suggesting that SSRP1 plays a crucial role in microtubule growth and spindle assembly during mitosis [21]. However, in this study there was no statistically significant interaction or regulatory pathway in which SH3RF2 is involved during myotube differentiation (Figure 5B). In this study, we analyzed global RNA sequencing data focusing on muscle differentiation pathway and so, it would be possible that SH3RF2 plays an alternative role(s) in other biofunction(s). Alternatively, Rubin et al [16] reported SH3RF2 expression in brain and muscle and it could be also possible that SH3RF2 is more importantly working in brain than muscle. It will be important to determine the roles of global regulatory and/or signaling pathways for various biological processes in the future. Additionally, in vivo validation through the generation of CRISPR/Cas9-mediated SH3RF2 KO chickens should improve our understanding of the biofunction of SH3RF2. The advantages of the CRISPR/Cas9 technical platform are its efficient and precise genetic changes. Thus, the CRISPR/Cas9 technical platform facilitates functional analyses of specific genes in avian cell lines. Furthermore, the generation of genome-edited poultry mediated by the CRISPR/Cas9 system can be widely applied to the agricultural industry.




 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print







