The complete mitochondrial genome sequence of the indigenous I pig (Sus scrofa) in Vietnam
Article information
Abstract
Objective
The I pig is a long nurtured longstanding breed in Vietnam, and contains excellent indigenous genetic resources. However, after 1970s, I pig breeds have become a small population because of decreasing farming areas and increasing pressure from foreign breeds with a high growth rate. Thus, there is now the risk of the disappearance of the I pigs breed. The aim of this study was to focus on classifying and identifying the I pig genetic origin and supplying molecular makers for conservation activities.
Methods
This study sequenced the complete mitochondrial genome and used the sequencing result to analyze the phylogenetic relationship of I pig with Asian and European domestic pigs and wild boars. The full sequence was annotated and predicted the secondary tRNA.
Results
The total length of I pig mitochondrial genome (accession number KX094894) was 16,731 base pairs, comprised two rRNA (12S and 16S), 22 tRNA and 13 mRNA genes. The annotation structures were not different from other pig breeds. Some component indexes as AT content, GC, and AT skew were counted, in which AT content (60.09%) was smaller than other pigs. We built the phylogenetic trees from full sequence and D loop sequence using Bayesian method. The result showed that I pig, Banna mini, wild boar (WB) Vietnam and WB Hainan or WB Korea, WB Japan were a cluster. They were a group within the Asian clade distinct from Chinese pigs and other Asian breeds in both phylogenetic trees (0.0004 and 0.0057, respectively).
Conclusion
These results were similar to previous phylogenic study in Vietnamese pig and showed the genetic distinctness of I pig with other Asian domestic pigs.
INTRODUCTION
The I pig originated from Nam Dinh province, Vietnam and before 1970s it was common in Red River delta. I pig has some characteristics as black skin, short, sparse and black hair, big and short mouth, short body, sagging back, hanging down belly, ten breasts, and weak legs. There are several remarkable characteristics of I pig, e.g., they can adapt to poor quality feed, harsh climate, disease resistance, good quality meat, high nutrition and small string. This breed is early maturity (3 to 4 months for female and 6 to 8 months for male), from 110 to 115 days of pregnant period and births 8 to 10 offsprings in once. However, I pig size is quite small, the body weight at four months is less than 14 kg, mature pigs weight less than 70 kg and percentage of lean meat is less than 40%, so it is gradually replaced by Mong Cai pig, which has higher productivity. Nowadays, there are approximately 20 individuals locate in Thanh Hoa province [1,2]. In 2005, Vietnam Ministry of Agriculture and Rural Development put I pig on the list of rare breeding animal genetic resources need to be conserved and also showed the importance of I pig genetic resources.
The characteristic mitochondrial DNA (mtDNA) structure and function is suitable to become a molecular maker to evaluate the molecular evolution, classification, population genetic analysis, and relationship identification. Firstly, mtDNA is a small, less than 20 kb circular sequence [3]. Secondly, it is haploid, maternally inherited and no recombination [4]. Thirdly, the mutation is primarily single base pair substitutions [5] and the evolution rate of mtDNA is more rapid and constant than nuclear DNA [3,6]. Animal mtDNA has one control region and codes for 13 protein subunits, 2 rRNAs, and 22 tRNAs [7]. The control region as known as D loop, which regulates both replication and transcription of mtDNA, has used in several previous phylogenetic studies [8,9].
In the present study, we report the full sequence of I pig mitochondrial genome. The full sequence and D loop region sequence are used to estimate the genetic relationships between I pig and Asian and European pigs. The phylogenetic results are useful for classification and identification of genetic origin in I pig breed conservation activities and further studies.
MATERIALS AND METHODS
Sample collection and DNA extraction
All animals used for sample isolation were handled in accordance with the National Institute of Animal Sciences (NIAS) (Hanoi, Vietnam) care and management guidelines. Five milliliter (5 mL) of blood was collected with anticoagulant from jugular vein of the I pigs reared in the Thanh Hoa province (Vietnam) according to I pig breed conservation project of NIAS. Total DNA extraction was performed by standard phenol-chloroform method [10].
Amplification by polymerase chain reaction
The full sequence of mtDNA was amplified by using 30 primer pairs into 30 overlap segments (Table 1). Each 25 μL polymerase chain reaction (PCR) reaction consist of 12.5 μL GoTaq Green Master Mix, 1.0 μL DNA template, 0.5 μL of each primer (10 ppm), and 10.5 μL sterile deionized water. The PCR thermocycler was an initial denaturation at 94°C for 5 min, followed by 25 cycles of 94°C for 30 s, 50°C to 55°C based on the primer sequences for 30 s, and 72°C for 50 min, with a final extension at 72°C for 10 min. The product sequence length was checked by 1% agarose gel electrophoresis. PCR products were purified by QIAquick PCR Purification Kit (Qiagen, Hilden, Germany).
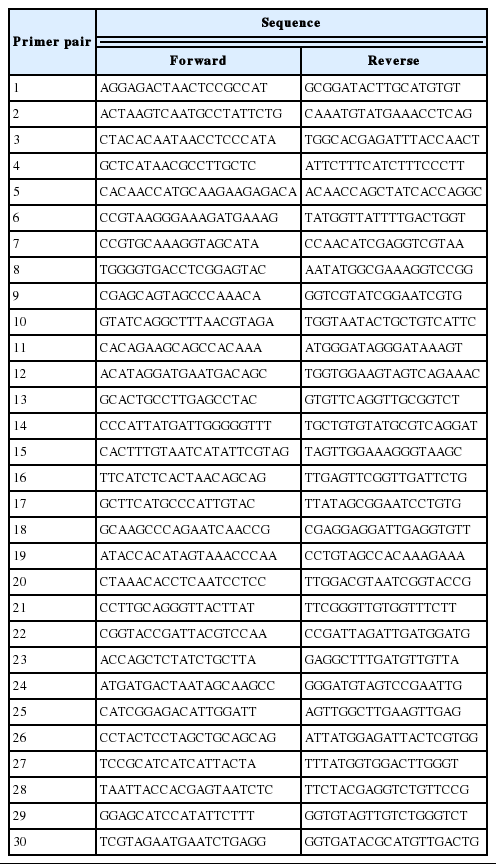
Primer pairs for I pig complete mitochondrial DNA
Sequencing and structure analysis
The PCR products were sequenced using ABI3500 DNA sequencer. The DNA sequences were assembled using DNA Dragon v1.6.0 software and exported to FASTA file. The complete sequence was annotated using MITOS and DOGMA. The mitochondrial circle structure was built using GenomeVx and the tRNAs structure was downloaded from online report of MITOS analysis result. The C, G, A, T frequencies and A+T-content of the entire mitochondrial genome were calculated using DAMBE v6.3.17 [11]. After that, skewness for each protein coding gene was evaluated using the following formulas: GC skew = (G−C)/(G+C) and AT skew = (A−T)/(A+T).
Phylogenetic analysis
The D loop and full sequence of I pig were aligned with other pig sequences using MUSCLE algorithm [12] of MEGA6 [13]. The best model for full mitochondrial genome group was HKY+G and TN93+I. We used Bayesian phylogenetic analysis in BEAST v1.8.3 [14] with Yule process and MCMC 10000000 to calculate the alignment data and Figure Tree v1.4.2 to build the phylogenetic trees.
RESULTS
Gene annotation
The total length of I pig mitochondrial genome was 16,731 base pairs (bp) (Figure 1). Similar to other Sus scrofa mitogenomes [15,16], there were 2 ribosome RNA genes, 13 protein coding genes, 22 transfer RNA genes and a non-coding D loop region. The base composition was 34.66% for A, 26.24% for C, 13.35%, for G and 25.75% for T, the A+T content was 60.41%. The D loop region was 1,295 bp in length and located between tRNA-Phe and tRNA-Pro. In addition, there were 12 other non-coding spaces in the length of 1 to 32 bp and 8 overlaps in the length of 1 to 43 bp among all genes. The length of 12S and 16S rRNA was 960 and 1,570 bp, respectively. Twenty two transfer RNA genes were range from 59 bp to 75 bp in size. The total length of 13 protein coding genes was 11,413 bp ranging from 204 bp to 1,821 bp. In which, four genes (cytochrome c oxidase subunits [COX] 2, COX3, nicotinamide adenine dinucleotide dehydrogenase subunits [ND] 3, and ND4) had incomplete stop codon T--, that presumptively formed a complete stop codon by post-transcriptional polyadenylation. The start and stop of all coding genes were same as other pig breeds. The GC and AT skews showed the nucleotide composition bias in the mitogenome sequence. All protein and rRNA coding genes located in H strand exhibited negative GC skew and positive AT skew, this reflected the bias of Cytosine and Adenine. In the H strand, the most Cytosine bias was adenosine triphosphatase subunits (ATPase) 8 (−0.55), the less Cytosine bias was 16S rRNA (−0.11), whereas 12S rRNA gene had the most Adenine bias and COX1 gene had Cytosine content equal Guanine. The only one gene coded in L strand was ND6 had positive GC skew (0.56) and negative AT skew (−0.35) (Table 2). Because the tandem repeat motif “CGTGCGTACA” could cause heteroplasmy [17], we removed them out the mitogenome sequence before analysis. The component of I pig full sequence after remove motif was 34.83%, 26.19%, 13.15%, and 25.82% for A, C, G, and T, respectively. Compared the AT content, GC and AT skew between I pig sequence and other pig breeds, the range of AT content, GC skew and AT skew of published full sequence were (60.19; 60.82), (−0.34; −0.30) and (0.12; 0.15), respectively. The range of AT content, GC skew, and AT skew of published D loop sequence were (60.73; 61.80), (−0.38; −0.31); (0.15; 0.19), respectively. We found that the AT content of I pig D loop (60.09%) was smaller than other pigs, whereas its full sequence (60.65%) was larger than Bihu pig (60.19%). The D loop of I pig also had less Cytosine bias than most pig breeds, excepted WB Yunnan (−0.31). GC and AT skew of full genome and AT skew of D loop did not have too much different (Table 3).
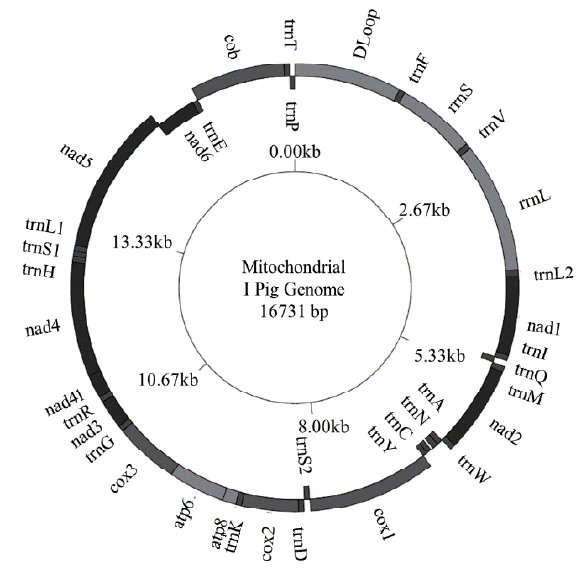
The circular map of the I pig mitogenome used GenomeVx.

Mitochondrial genome organization of the I pig
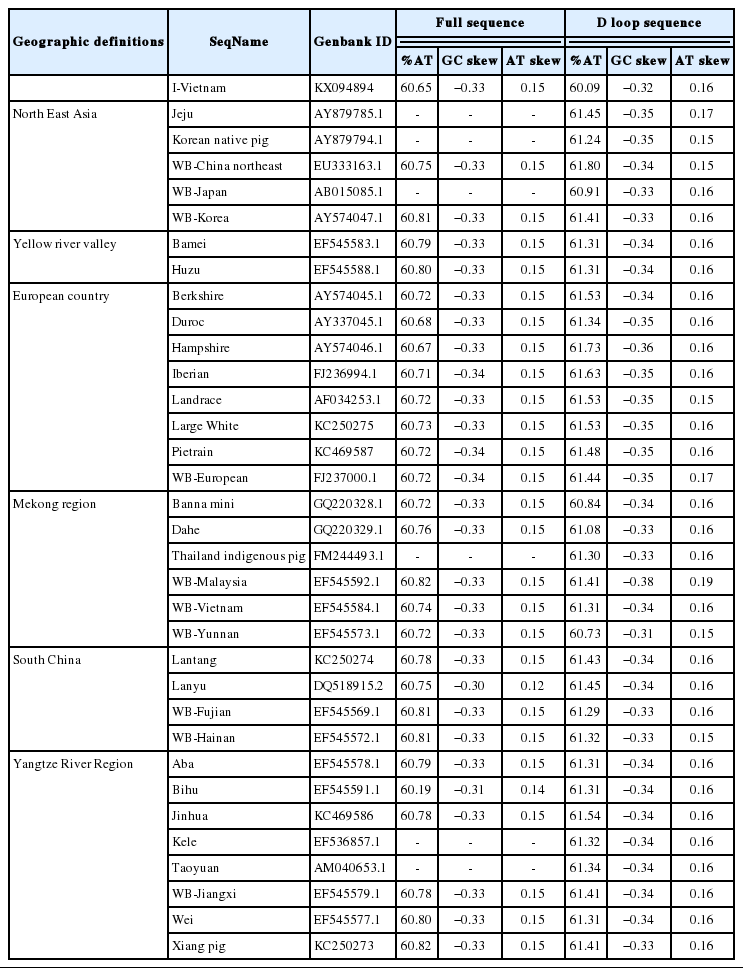
Pig breeds, their geographic definitions and sequence components
The secondary structure of tRNAs was predicted by MITOS online software (Figure 2). Twenty one tRNAs excepted for tRNA Ser-1 had the typical cloverleaf structure. The tRNA Ser-1 dihydrouridine arm did not form a stable structure.

Secondary structure of 22 tRNAs encoded by the I pig mitogenome.
Phylogenetic analysis
We built the phylogenetic trees of the I pig full sequence and D loop sequence with the published mitogenomes of pig breeds from three different geographical regions: European Countries, Mekong region and China. In both phylogenetic trees, most of the pigs divided into two clades: Asian clade and European clade, the genetic distances between them in full sequence and D loop sequence phylogenetic trees were 0.008 and 0.0137, respectively.
The twenty-seven public reference sequences were used to build the full sequence phylogenetic tree (Figure 3). There were two large groups in the Asian clade. Although the Berkshire and WB Korea were in Asian clade they felt outside of both large groups, the distance from WB Korea was 0.0045 and Berkshire was further (0.0053). Genetic distance between two groups was 0.0004. The I pig, Banna mini pig, and WB Vietnam and WB Hainan clustered a group. I pig was a sister taxon with the Banna mini (0.0002), close was the WB Vietnam (0.0016) while the WB Hainan had most distant relationship (0.0022).
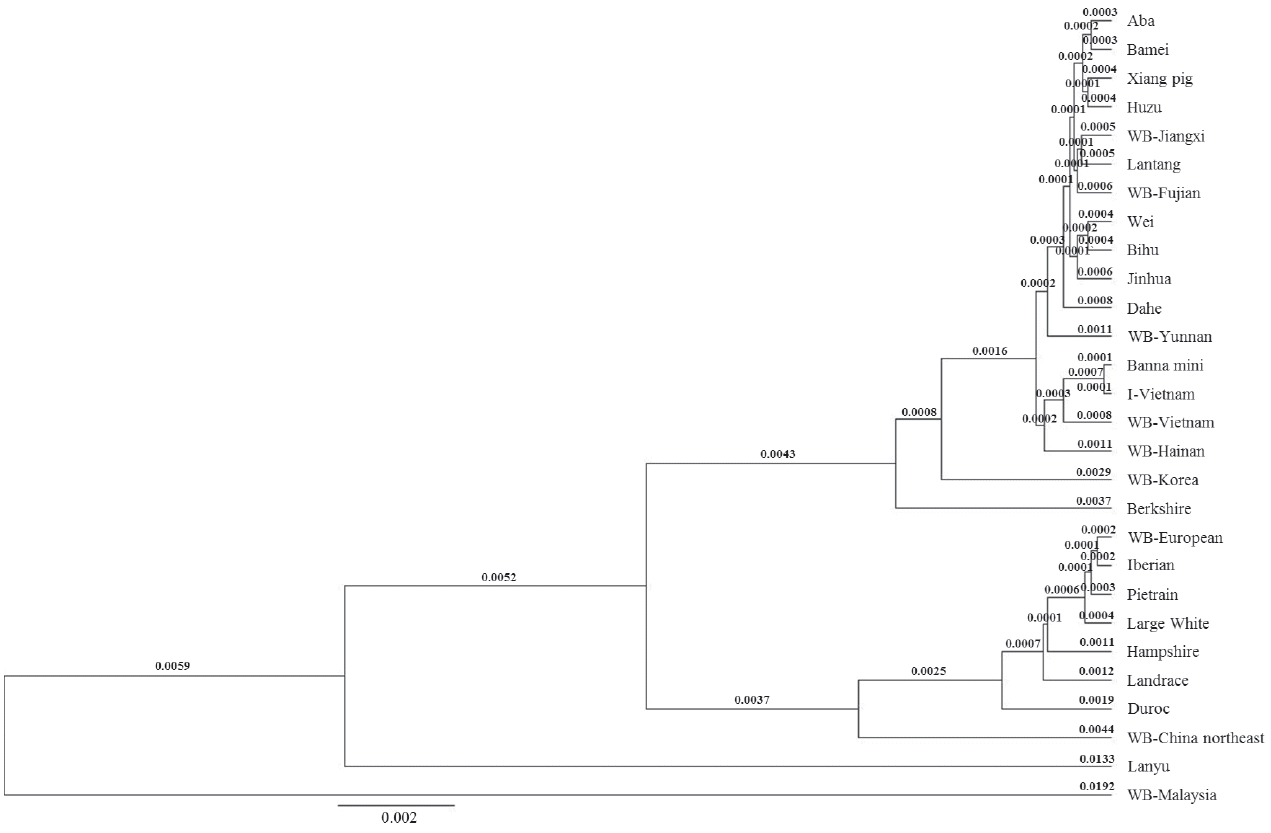
Full sequence phylogenetic tree using Bayesian method. WB, wild boar.
There were 33 sequences in the D loop phylogenetic tree and this tree showed the different distribution (Figure 4). The WB Japan and WB Korea joined to the group of I pig. Therefore, this group could be separated into two subgroups: I pig and Banna mini continued to be cluster (0.0006) and WB Vietnam, WB Korea and WB Japan were another subgroup. The genetic distance between the two subgroups was 0.0065. WB Hainan felt out of this group, it and Taoyuan pig became sister taxons (0.0014). The genetic distance between the two big groups of the Asian clade was 0.0057.
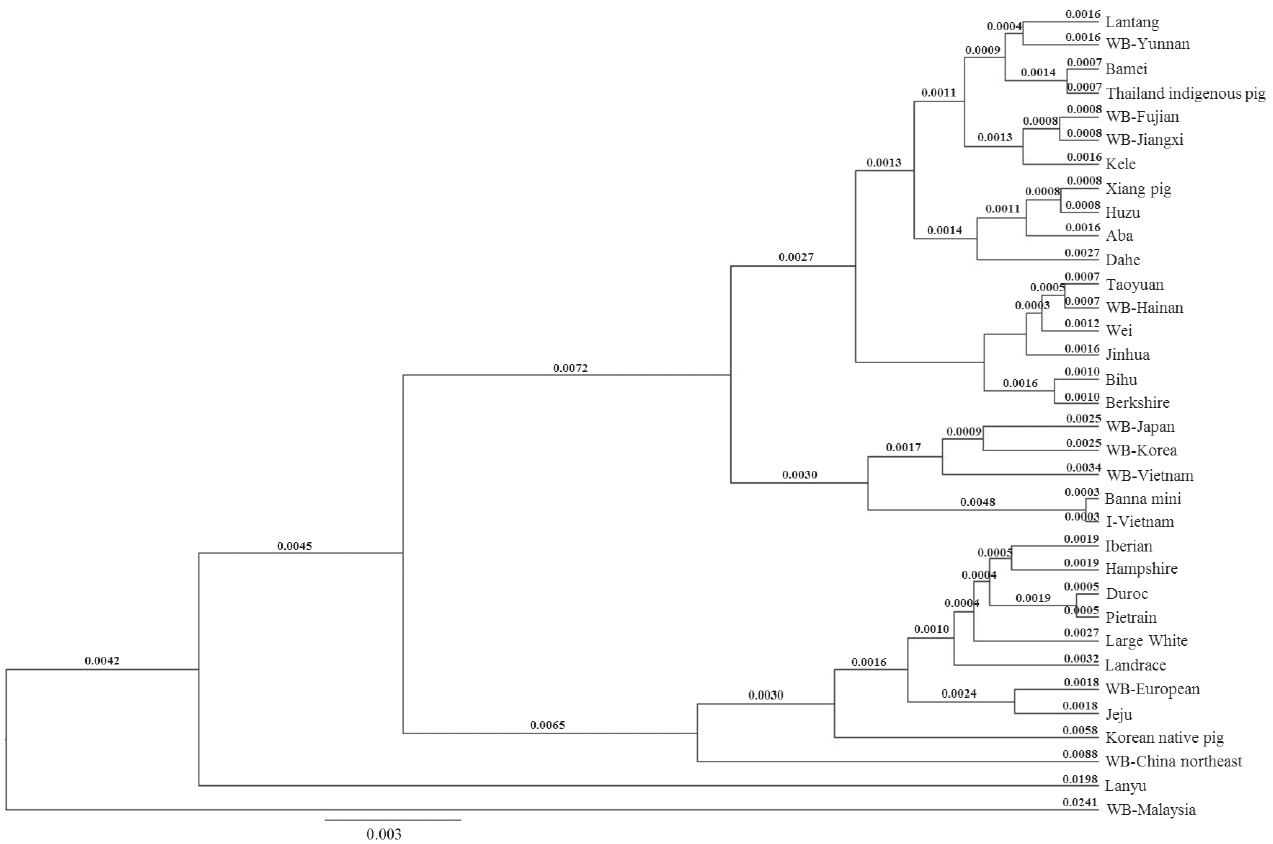
D loop sequence phylogenetic tree using Bayesian method. WB, wild boar.
DISCUSSION
This study was the first molecular research in I pigs. The result produced an overview of I pig mitogenome and compared it with other published Sus scrofa mitochondrial sequences. Although the I pig mitochondrial structure was similar to the pig breed mitochondrial structure in previous researches, there were some differences in sequence components of I pig especially in D loop region. AT content, GC skew, and AT skew were used to study the nucleotide-compositional behavior of mitochondrial genomes, and related to phylogenetic [18,19]. So that, the difference of I pig component indexes showed the genetic variation of I pig with pig breeds taxa.
The phylogenetic trees showed the difference between Asian and European pigs. This result identified the divergence point of Asian and European domestic pigs had occured [20] or 746,000 years before present [21]. And, phylogenetic trees illustrated the contribution of Asian and European pig breeds to the development of each other, which were similar to the genetic evidence Giuffra found for introgression in the late 18th and early 19th centuries [20].
The previous researches reported the close genetic distance between Vietnamese pig and Asian pig. Both of phylogenetic trees in our study showed similar results, namely I pig had a close genetic relationship with Banna mini, a Mekong pig breed, WB Vietnam and other Asian wild boars. While the most of Chinese pig and some breeds of the Mekong region and Berkshire pig became a separate large group. This result agreed with a study of Lan and Shi which showed that Vietnamese wild boar and Chinese domestic pig were separated by a large genetic distance [22]. Moreover, a later study of Hongo et al [23] compared the 574 bp mitochondrial DNA sequences of 30 Vietnamese pigs with Asian domestic pigs and wild boars revealed some Vietnamese domestic pigs were closely related to wild boars and genetically distinct from East Asian domestic pig groups. However, there are a need for additional studies to ensure the domestic origin of I pig in Vietnam.
In present research, we reported the mitochondrial full genome sequence of I pig (KX094894) in NCBI genbank. The sequence was used to predict and analyze the component and structure of I pig mitogenome. The result of the phylogenetic analysis was conducted to illustrate the genetic separation between I pig and most of Asian domestic pigs.
ACKNOWLEDGMENTS
This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number: 106-NN.05-2014.66.
Notes
CONFLICT OF INTEREST
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.