



INTRODUCTION
The identification of the source of meat is a sensitive and important issue concerning food products especially in Moslem countries. The food safety and assurance consists some aspects including health, hygienic and halal authentication. An effective method for detecting pork in food from meat or non-meat products is essential to avoid falsification. The falsification of food products including the meat products and non-meat products is becoming common due to a higher profit return by mixing halal meat with non halal meat.
A reliable, sensitive and exact procedure for the authentication is crucial for law enforcement, quality assurance and consumer protection. Authenticity testing and analytical techniques have improved immeasurably recently and now can draw on a wide variety of techniques and methods, each appropriate and specific to deal with a particular problem (Ballin, 2010). Several different techniques are currently used for determination of species, primarily relying on detecting differences in proteins and are either electrophoretic or immunological methods. One of the major drawbacks with such methods is that they do not perform well with heat treated samples. More recently DNA techniques have been applied to meat specification with advantage that the DNA is much more stable at cooking temperatures.
Polymerase chain reaction (PCR)-based assays are currently the method of choice for species identification. PCR analysis of species-specific mitochondrial DNA sequences is the most common method currently used for identification of meat species in food and seems to be best as a ‘routine’ test, because it is easy, rapid and allows the discrimination of several species at the same time (Bottero and Dalmasso, 2010). Among the PCR based methods PCR-restriction fragment length polymorphism (RFLP) using various genes is one of the options which can be applied for species differentiation. The PCR-RFLP procedures have been used for species identification by exploiting DNA sequence variation within the mitochondrial DNA (mtDNA) including the mt-12S rRNA (Fajardo et al., 2006) and 16S rDNA and nuclear markers (Rastogi et al., 2007). Sun and Lin (2003) have applied the PCR-RFLP analytical method based on mitochondrial 12S rRNA for identifying porcine, caprine and bovine meats. A similar method using 12S rRNA PCR-RFLP was also applied to the identification of beef, buffalo and chevon meats (Girish et al., 2005).
The advantage of mitochondrial-based DNA analyses derives from the fact that there are many mitochondria per cell and many mitochondrial DNA molecules within each mitochondrion, making mitochondrial DNA a naturally amplified source of genetic variation (Unajak et al., 2011). Additionally, procedures like polymerase chain reaction mean that the sensitivity of these test are greatly enhanced and positive reaction are possible from the merest trace of the species under investigation. Erwanto et al. (2012) also developed PCR-RFLP methods based on mitochodrial DNA using BseDI restriction enzyme for meat species differention and were able to distinguish the presence of pork in meatballs on a laboratory scale which could detect 0.1% level of pork.
However the traceability and identification of pork and other derivate porcine contamination in most popular foods in Indonesia has not been previously done. Therefore, this study was aimed at detection of pork or derivative porcine contamination in commercial product of meatballs from Surabaya and Yogyakarta region of Indonesia using PCR-RFLP methods through cytochrome b gene.
MATERIALS AND METHODS
Sample preparation and DNA extraction
Meatballs were obtained from thirty nine different meatball shops in the Surabaya and Yogyakarta region. DNA was extracted and purified from meatball samples using Sambrook et al. (1989) methods with slight modification. Approximately 50 mg of meatballs were blended using a commercial blender and placed in a 1.5 mL micro centrifuge tube. A-500 μL of Tris-EDTA-NaCl (TEN) buffer, 10 μL Proteinase K and 50 μL 10% sodium dodecyl sulfate were added and mixed by vortexing. The mixture was incubated at 42°C in a water bath overnight to disperse the sample until the tissue was completely lysed. After overnight incubation, 50 μL 5 M NaCl, 40 μL phenol and 400 μL chloroform-isoamyl-alcohol was added to the mixture and then incubated at 37°C for 1 h and centrifuged at 3,000 rpm for 5 min. The supernatant was taken and put in a new eppendorf tube and mixed by vortexing for 30 s, then 50 μL 5 M NaCl and 1 mL absolute ethanol were added, mixed vigorously and incubated at −20°C for 15 min. The DNA was precipitated from the mixture after centrifuging at 8,000 rpm for 5 min and the supernatant discarded. The ethanol was gently poured off and the pellet resuspended in 0.5 mL of Tris-boric acid (TB) buffer. A 0.3 mL of 5 M ammonium acetate and 1 mL of 95% to 100% ethanol were added to the resuspended DNA for a second precipitation. The solution was then placed at −20°C for 10 min. The tubes were then spun at top speed in a microcentrifuge and the ethanol supernatant poured off gently. The resulting pellet was resuspended in Tris-EDTA (TE) buffer and stored at −20°C for further analysis.
Polymerase chain reaction amplification of cytochrome 2b of mitochondrial gene
The set of primers used for amplification consisted of Cyt b-Forward (FW) and Cyt b-Reverse (REV) oligonucleotides as follows:
CYT b FW 5′-CCA TCC AAC ATC TCA GCA TGA TGA AA-3′
CYT b REV 5′-GCC CCT CAG AAT GAT ATT TGT CCT CA-3′
Amplification of the mt cyt b gene was performed in a final volume of 25 μL containing 250 ng of extracted DNA, mega-mix royal (optimized mixture of Taq polymerase, anti-Taq polymerase monoclonal antibodies in 2×reaction buffer (6 mM MgCl2 with 400 μM dNTPs, stabilizer and blue loading dye) (Microzone Ltd, West Sussex, UK) and 20 pmol of each primer. Amplification was performed with a thermal cycler according to the following PCR step-cycle program: Pre-denaturation at 94°C for 2 min to completely denature the DNA template, followed by 35 cycles of denaturation at 95°C for 36 s, annealing at 51°C for 73 s. and extension at 72°C for 84 s. Final extension at 72°C for 3 min followed the final cycle for complete synthesis of elongated DNA molecules. Two microliters of PCR products were electrophoresed at constant voltage (50 V) on 2% agarose gel (Promega, Madison, WI, USA) for about an hour in 1× TBE buffer, pH 8.0 and stained by ethidium bromide. A-100 bp DNA ladder (Promega, USA) was used as size reference. The gel photo was taken using the Syngene gel documentation system.
Restriction fragment length polymorphism
Two units/μL of RE BseDI (Fermentas) were applied to 10 μL of amplified DNA in a final volume of 20 μL digestion mixture [containing 1× reaction buffer (10 mM Tris-HCl, 100 mM KCl, 1 mM EDTA, 0.2 mg/mL BSA, 1 mM DTT and 50% glycerol)] and were incubated at 55°C for 3 h for optimal results. Five μL of the digested samples were electrophoresed at constant voltage (50 V) on 2% agarose gel (Promega, USA) for about an hour in 1× TBE buffer, pH 8.0 and stained by ethidium bromide. A-100 bp (Promega, USA) was used as size reference marker. The gel photo was taken using the Syngene gel documentation system.
RESULTS AND DISCUSSION
The consumption of meat in developing countries is rising year by year and followed by an increasing demand for protection from falsely labeled food for religious and health reasons. People are requesting researchers to provide a system to detect contamination of beef with pork because some contamination is done for the economic reason that beef is more expensive than other meat, especially pork. The present study aimed to apply a PCR-RFLP method using cytochrome b gene that has already confirmed for pork detection in meatball products on a laboratory scale (Erwanto et al., 2012) to the specific identification of pork in meatballs from commercial meatball shops in Indonesian local markets.
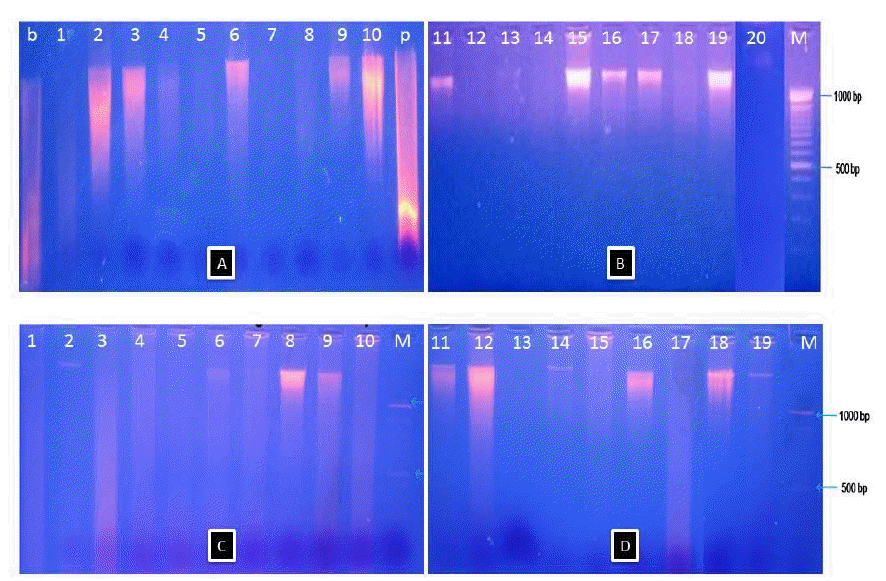
Genomic DNA was used as template for amplification by PCR with the universal primer. The first step of the protocol for PCR analysis is the extraction of genomic DNA. The extraction genomic DNA from the commercial meatballs samples was reliable although there were some smearing of the DNA in the samples. Figure 1 showed the electrophoretic patterns of genomic DNA from meatballs of Indonesia local market.
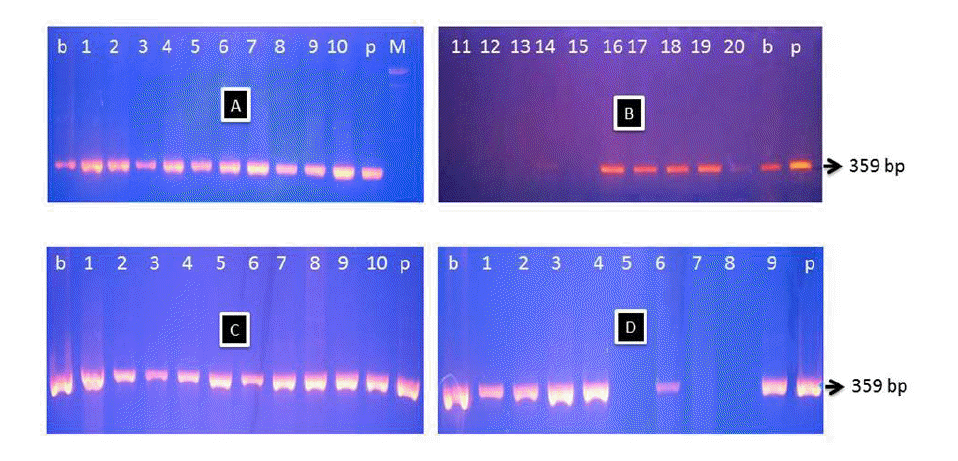
The PCR has the potential sensitivity and specificity to attain detection of a target sequence from template DNA. The PCR products of the cytochrome b gene as an amplicon from the meatball DNA from thirty nine locations of meatball shops are shown in Figure 2. The cytochrome b gene amplified by PCR from a meatball after boiling resulted in a DNA fragment of approximately 360 bp. These small amplicons are ideal for use with processed foods where DNA is commonly degraded. This result indicated that both pig and beef DNA in processed meat was successfully amplified in the PCR reaction.
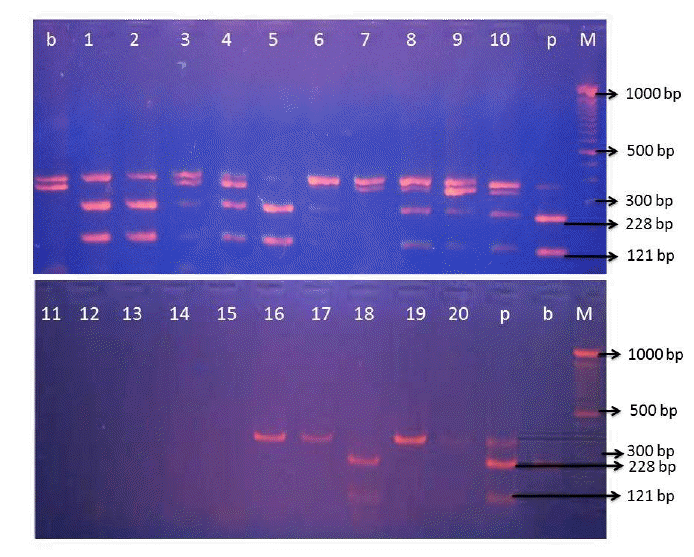
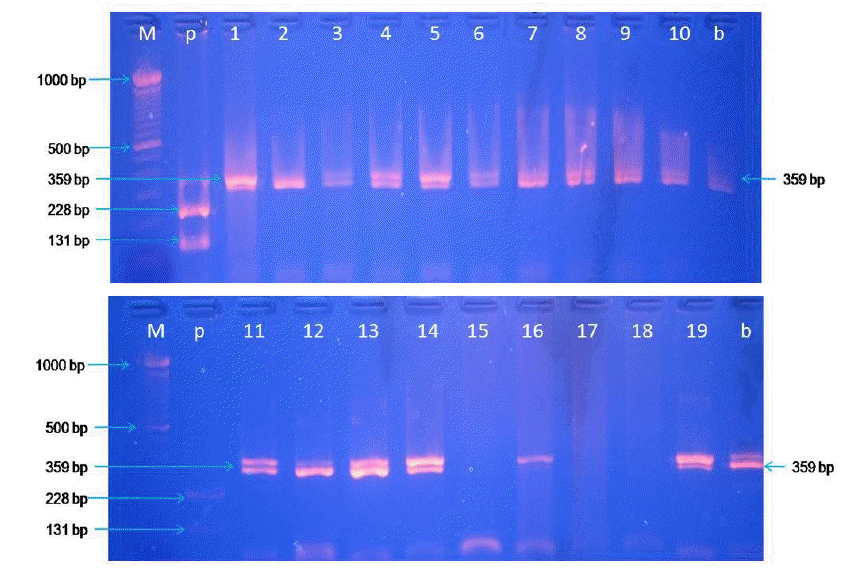
The PCR product was digested using BseDI restriction enzyme at 55°C for 3 h. The result of RFLP analysis of meatball samples from various shops in Yogyakarta region (Figure 3) indicated some positive pork contamination. Erwanto et al. (2011) reported that cyt b gene from pig species could be digested by BseDI restriction enzyme and 0.1% contamination level of pork could be detected in sausage products using this method. Data indicated pork contamination from nine meatball shops in Yogyakarta region. The contamination by pork was not found in meatballs from various meatball shops in Surabaya region (Figure 4). BseDI cleavage bands visualized in the gel were suitable for the discrimination of pork in processed food. BseDI end nuclease cleaved the cytochrome b gene products of pig species into two DNA fragments of 228 and 131 bp (Figure 3; lane 1, 2, 3, 4, 5, 8, 9, 10, and l8).
The nine positive pork contamination samples from the Yogyakarta region were an unpredicted and unexpected result of this research because almost meatball shops declared only beef on the ingredients label and only one meatball shop stated that they were using pork meat. The Moslem population in Yogyakarta region is around 3 million (90%) from the total population and pork meat is prohibited for them. Although there was clearly pork contamination, this finding could not exactly conclude that the pork contamination was deliberate as contamination could have occurred during processing especially in the grinding process. In Yogyakarta many meatball industries obtained meat from the traditional market and were ground using the traditional equipment which also may be used for pork while the meatball producer in Surabaya may have been using private equipment just for beef meat.
The PCR-RFLP approach based on mitochondrial DNA polymorphism has already been established for species identification of common farm animals (Chen et al., 2012). This result also indicated that meatballs which are commonly cooked for more than 30 min did not degrade DNA into a small size (<200 bp). Tanabe et al. (2007) investigated the feasibility of using mitochondrial cyt b gene to detect porcine DNA in commercial food products from a market. Electrophoresis of the PCR product clearly detected porcine DNA, while no amplification occurred in others meat sources: cattle, chicken, sheep, and horse.
The optimum PCR conditions are affected by some factors including the temperature, design primer, polymerase enzyme and also buffer quality. Several analytical methods using PCR have been developed to qualitatively detect the presence of non halal meat species or commercial fraud of ingredients in the food industry. Unajak et al. (2011) used nested PCR analysis of mitocondrial DNA with species specific primers, Tanabe et al. (2007) PCR with cloning of target genes, Aida et al. (2005) using PCR-RFLP and He et al. (2013) developed new methods based on DNA in different refined oils and adulterated oils with an optimized extraction method and evaluated by nano real time PCR.
The BseDI enzyme cleaves the cytochrome b gene products of pig species into two DNA fragments of 228 and 131 bp (Erwanto et al., 2012). This research enabled finding that some beef meatballs in shops were contaminated with pork. The discovery of pork contamination in meatballs from the local market allows us to make some inferences. The first is that some meatball producers may have chopped the raw beef with the same machine that was used for pork. The second interpretation is meatball producers substituted pork for beef for economic reasons. Pork is cheaper than beef, consequently replacement of beef with pork increased profit.
Ong et al. (2007) used various restriction enzymes like AluI, BsaJI, and RsaI to identify pork from different mixtures of meat; chicken, pig and cattle in various modifications. Another study by Fajardo et al. (2006) reported PCR-RFLP based on the mitochondrial 12S rRNA fragment cannot be applied for species identification in thermally treated meats as the thermal action or other processing may degrade the DNA present in the food tissues. However the present result allows us to report that PCR-RFLP of the mitochondrial cytochrome b gene is a suitable alternative that can be applied to the detection of pork in processed meat in commercialized products such as meatball and sausage.
BseDI represent one of the restriction enzymes of type II which is able to recognize the nucleotide sequence and cut the DNA molecule specifically. BseDI is isoschizomer of BsaJI and SecI. This enzyme cuts the DNA molecule at sequence of C↓CNNGG. BseDI enzyme was obtained from bacterium of Geobacillus strearothermophilus (Fermentas AB) and Bacillus strearothermophilus RFL1434 (US, Biological), while BsaJI enzyme from bacterium of Geobacillus strearothermophilus J695 (New England Biolabs).
Our previous research showed that compared with BsaJI endonuclease profiles, the DNA restriction patterns obtained after digestion of the amplicons with BseDI enzymes consisted of same patterns. The difference between BsaJI and BseDI restriction enzyme is the incubation time for the digestion. BseDI needed 3 h for digestion (Erwanto et al., 2012), while BsaJI enzyme needed more than 12 h (Aida et al., 2005).
Halal food has been an important issue not only in predominately Moslem countries but also in many other countries. Halal food production is important not only for religious reasons but it also has an economic impact on many industries. Valid methods for detection of non halal ingredients in food products are required to answer the halal issue. The PCR-RFLP method was fast and straightforward and it might be a useful tool in the quality control for pork detection in food products.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print







