



INTRODUCTION
MicroRNAs (miRNAs) are a class of endogenous, small non-coding RNAs, which are widespread and evolutionarily high-conserved in eukaryotes (Bartel, 2004). Most of mature miRNAs play very important roles in post-transcriptional gene regulation by directing degradation of mRNAs or facilitating repression of targeted gene translation (Lee et al., 1993; Lai, 2002). With the development of next-generation sequencing technology and bioinformatics, more and more miRNAs have been identified in various organisms (Yousef et al., 2009; Li et al., 2011a), the number of miRNAs discovered and deposited in miRBase 18.0 (http://www.mirbase.org/) (Kozomara and Griffiths-Jones, 2011) has increased approximately exponentially. It is estimated that number of miRNA might account for 2 to 5% of the total number of all mammalian genes and collectively regulate up to 60% of protein-coding genes (Lewis et al., 2003; Berezikov et al., 2005), therefore, it is likely that the identification of all animal miRNAs is far from complete. For example, the number of mature miRNAs identified from domesticated ruminants, such as Bos taurus and Ovis aries is 676 and 103 mature miRNAs, respectively, which is far less than those identified from Homo sapiens or Mus musculus. Unfortunately, no miRNAs from Capra hircus have been deposited in the miRBase 18.0.
To expand the repertoire of miRNAs, we constructed three cDNA libraries prepared from lactating mammary gland tissues at different lactation stages, each library was sequenced individually using Illumina/Solexa Genome Sequencer. The sequencing data were analyzed at BGI (Beijing Genomics Institute) using a bioinformatics pipeline and other publicly available prediction tools to identify the potential novel miRNAs candidates. We also conducted a detailed analysis for these putative novel miRNAs candidates and their target genes. GO (Gene Ontology, http://geneontology.org/) (Carbon et al., 2009) and KEGG (Kegg Kyoto Encyclopedia of Genes and Genomes, http://www.genome.jp/kegg/) (Minoru et al., 2012) databases were also used to annotate the enriched functions and pathways of target genes. As a result, we obtained 50 putative novel miRNAs, 55,448 target genes candidates were predicted, GO annotation and KEGG pathway analyses showed that 18.22% of target genes involved in the metabolic pathways, more than 46% of the target genes were annotated involving in biological processes. Our results expanded the repertoire of miRNAs, and provided useful information for the investigation into the spatial-temporal regulation functions of miRNAs in goats and other animals.
MATERIALS AND METHODS
Sample collection and preparation
Laoshan dairy goat, one of four Chinese native dairy goat breeds, was developed from Sannens dairy goat. Sannens dairy goat was first introduced to Laoshan by German preachers early in 1904, and used for crossing with local does from 1919. Currently, Laoshan dairy goat is an important domestic animal in the Chinese agricultural industry due to its use in milk and meat production, which is mainly distributed in the eastern part of the Shandong Province, China.
The mammary gland tissues from five 4-year-old Laoshan dairy goats were collected by surgery at three lactation periods (30 d postpartum (early lactation), 90 d postpartum (peak lactation), 210 d postpartum (late lactation)), respectively, and were frozen immediately in liquid nitrogen. Total RNAs were individually extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions, and stored at −80°C for further analysis. All experimental procedures were approved by the Ethics Committee of Laboratory Animals of Shandong Agricultural University.
Small RNA library construction and sequencing
Total RNAs from the same lactation period were homogenized and used for library construction and Solexa sequencing. Total RNA integrity was confirmed using an Agilent 2100 Bioanalyzer (Agilent, USA).
Small RNA library construction was conducted according to the procedure as previously described (Ji, 2012). Briefly, small RNA fragments of 18 to 30 were isolated and purified, the small RNAs ligated with 3′ and 5′ adaptors were used for RT-PCR amplification, and the cDNA was used for further amplification. The PCR products were purified using 10% PAGE and used directly for library construction and Solexa sequencing at the Beijing Genomics Institute (BGI), Shenzhen.
The original image data obtained by the Solexa deep-sequencing analyzer were automatically transformed into raw reads using base calling. After removing the low-quality reads and contaminated reads, the clean reads were obtained and were used for further analysis. First of all, these sequences were mapped to the Ovis aries genome using SOAP v1.11 software (Li et al., 2008) to analyze their expression and distribution on the genome. Then, the clean reads were compared against the non-coding (ncRNAs) (rRNAs, tRNAs, snRNAs, and snoRNA) deposited in the NCBI GenBank and the Rfam databases using BLAST to annotate the small RNA sequences. The unannotated sequences were used to predict potential novel miRNA candidates and their target genes by blasting with all animal miRNAs deposited in miRBase 18.0. Mireap v0.2 software (http://sourceforge.net/projects/mireap/) (Li et al., 2012) was used to predict novel miRNAs according to the following parameters: i) minimal miRNA reference sequence length of 20 nt and maximal miRNA reference sequence length of 24 nt; ii) minimal depth of Drosha/Dicer cutting site is 3; iii) maximal copy number of miRNAs on the reference is 20; iv) maximal free energy allowed for a miRNA precursor is −20 kcal/mol; v) maximal space between the miRNA and miRNA* is 35; vi) minimal base pairs of the miRNA and miRNA* is 14; vii) maximal bulge of the miRNA and miRNA* is 4; viii) maximal asymmetry of the miRNA/miRNA* duplex, 5; and ix) flank sequence length of miRNA precursor, 100. The selected sequences were then folded into a secondary structure using an RNA folding program, RNAfold software (http://rna.tbi.univie.ac.at/gi-bin/RNAfold.cgi). Finally, the MiPred (http://www.bioinf.seu.edu.cn/miRNA/) was used to filter out pseudo pre-miRNAs using the following settings: Minimum free energy >−20 kcal/mol or p-value >0.05 (Jiang et al., 2007).
Target prediction for novel miRNAs
We searched the goat EST database at NCBI (http://www.ncbi.nlm.nih.gov/sites/entrez?db=nucest&cmd=Search&dopt=DocSum&term=txid9925%5BOrganism%3Anoexp%5D) using Mireap software to identify the potential target genes for novel miRNAs according to the previously reported criteria (Allen et al., 2005; Schwab et al., 2005). Briefly, i) no more than four mismatches between the small RNA and the target (G-U bases count as 0.5 mismatches); ii) no more than two adjacent mismatches in the miRNA/target duplex; iii) no adjacent mismatches in positions 2 to 12 of the miRNA/target duplex (5′ of miRNAs); iv) no mismatches in positions 10 to 11 of the miRNA/target duplex; v) no more than 2.5 mismatches in positions 1 to 12 of the miRNA/target duplex (5′ of miRNAs); and vi) minimum free energy (MFE) of the miRNA/target duplex should be ≥75% of the MFE of the miRNA bound to its perfect complement.
GO enrichment and KEGG pathway analyses for potential target genes candidates
GO (Gene Ontology, http://geneontology.org/) database was used to enrich gene functions from three ontologies: molecular function, cellular component and biological process. The GO terms with adjusted p-values ≤0.5 are defined as significantly enriched in the target gene candidates. KEGG (Kegg Kyoto Encyclopedia of Genes and Genomes, http://www.genome.jp/kegg/) database was also used to annotate the pathways for target genes. The FDR ≤0.5 were considered as significance.
RESULTS
Overview of small RNA library sequencing
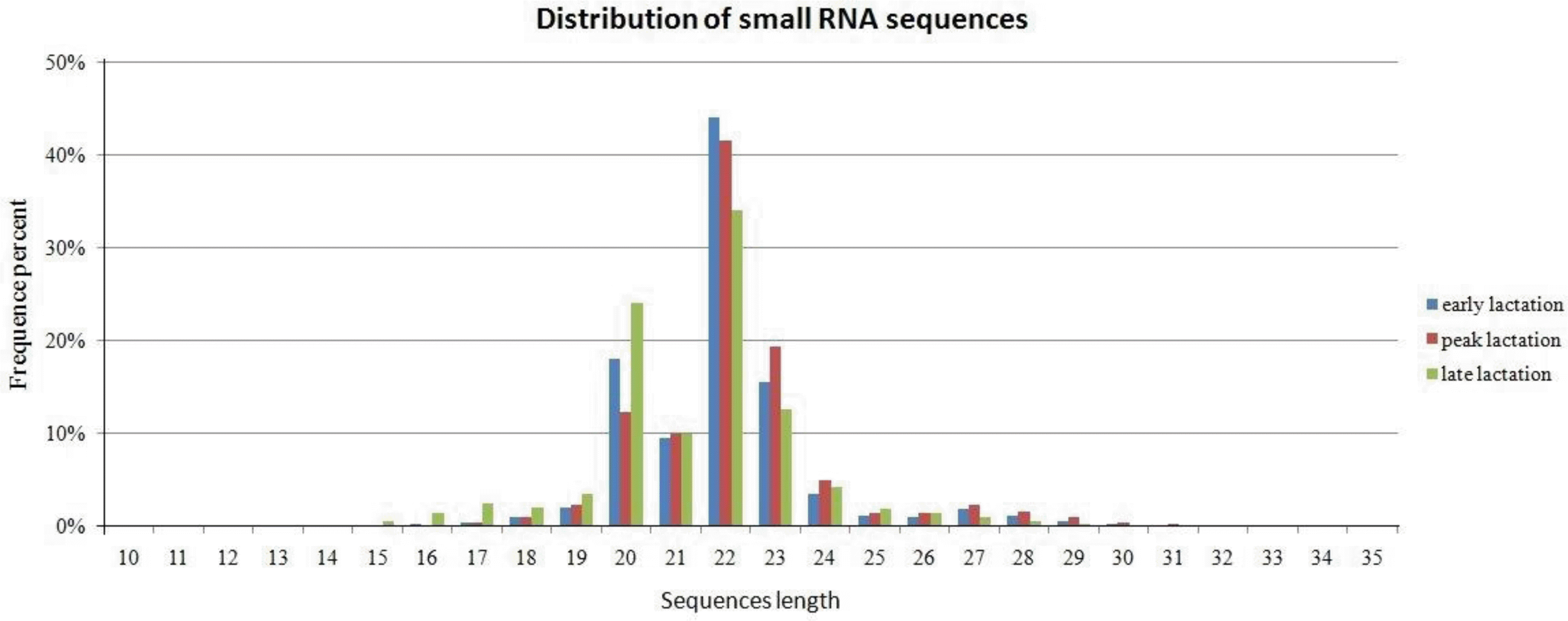
The sequencing reads for all three libraries were combined for simplicity and a unique list of sequence tags was generated with the corresponding raw abundance read counts. We obtained more than 40 million clean reads, ranging from 18 to 30 nt in length, the amount of data obtained using Solexa sequencing covered almost all small RNAs. As seen in Figure 1, the highest abundance was found for sequences with 22 nt, followed by 20 nt and 23 nt. The length distribution showed that more than 90% of the small RNA sequences were primarily distributed in 19 to 24 nt, which is consistent with the typical size of mature mammalian miRNA from Dicer digestion products (Zhang et al., 2009). The results were consistent with those in previous studies (Li et al., 2011a; Li et al., 2011b). After removal of low quality reads and adapter contaminants, 21,950,988 base sequences were perfectly mapped to Ovis aries genome, 2,673,840 reads were unannotated and remained for further novel miRNA candidates analysis (Table 1).
Novel miRNAs identification and target genes prediction
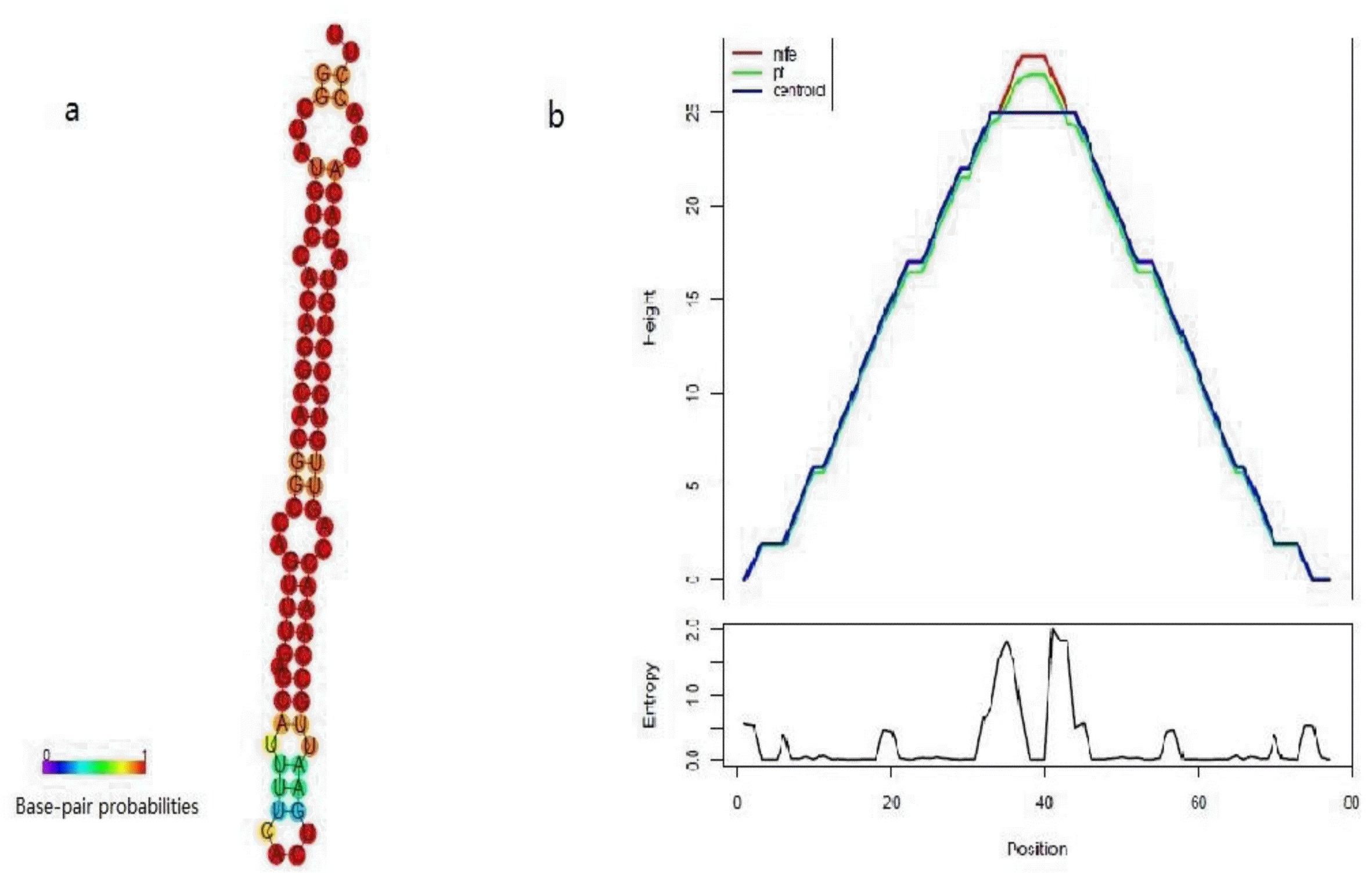
In this study, 2,673,840 unannotated small RNAs, matched to the Ovis aries genome, were used to predict potential novel miRNA candidates. After blasting to all animal miRNAs deposited in miRBase 18.0, a total of 50 potential novel miRNA candidates were predicted by bioinformatics. All of these new miRNAs were named temporarily in the form of Novel_miR_number, e.g., Novel_miR_1. Of these, only Novel_miR_7 was discovered in three libraries, 10 novel miRNAs were discovered in two libraries, 39 novel miRNAs were discovered in one library. All precursors of predicted novel miRNAs had regular stem-loop structures, with lengths ranging from 20 to 24 nt and reads ranging from 5 to 399, and had free energy ranging from −21.63 Kcal/mol to −66.70 Kcal/mol (with a average of −35.82 Kcal/mol). The predicted stem-loop structures of the 10 most abundant candidate miRNAs are shown in Table 2. Figure 2 shows a typical example of Novel-miR-7 secondary structure predicted using RNAfold software.
To understand the function of the newly identified miRNAs, the putative target genes of these miRNAs were predicted using Mireap software based on miRNA/mRNA interactions with default parameters (Materials and methods). A total of 55,448 annotated mRNA transcripts were predicted as putative target genes candidates for 50 novel miRNAs.
Go enrichment and KEGG pathway analyses
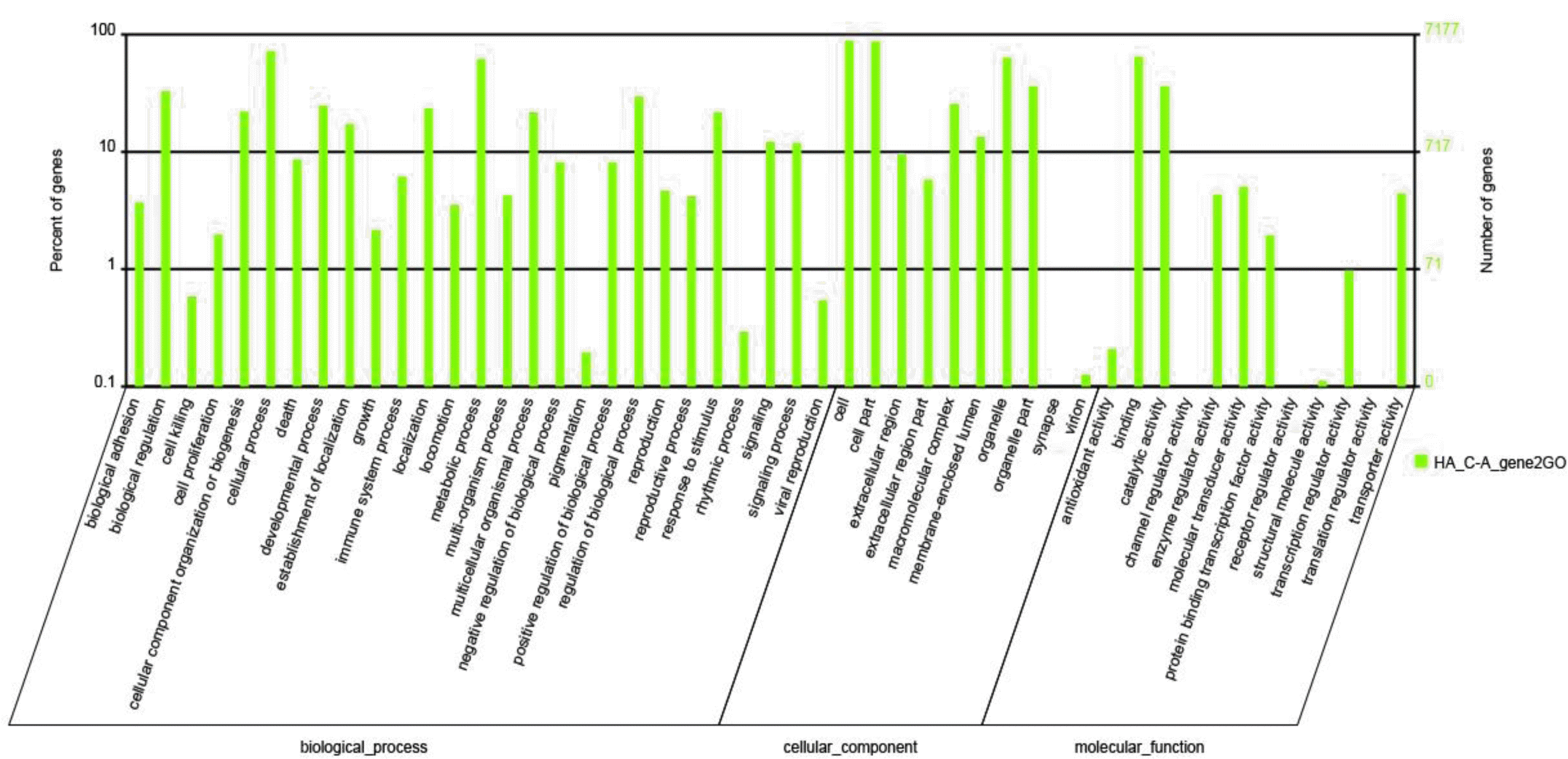
To further understand the physiological functions and biology processes involved by these miRNAs, those putative target genes were analyzed and annotated through GO and KEGG databases. The GO enrichment analysis revealed that more than 39% of the genes were involved in cellular component ontology, more than 46% of the genes in biological processes and approximately 14% of the genes in molecular functions (Figure 3). The KEGG analyses showed that approximately 18.22% of the genes were committed to a metabolic pathway, more than 15% of target genes involved in pathways associated with disease, such as Huntington’ disease and Alzheimer’ disease, about 8% of genes involved in the Ribosome pathway or Oxidative phosphorylation pathway (Figure 4).
DISCUSSION
miRNAs represent a new class of factors that can regulate gene expression. To date, many miRNAs have been discovered and deposited in miRBase 18.0, while there are numerous miRNAs that remain undiscovered. To identify these potential novel miRNAs, three cDNA libraries were built in the present study. An Illumina/Solexa high-throughput sequencer were used to obtain the high-quality sequencing data.
The Solexa high-throughput sequencer has a special advantage for discovering functionally important potential novel miRNAs that might not be detected using traditional Sanger sequencing, especially for the species without available whole genome information (Chen et al., 2009). In this study, through the use of Solexa sequencer, 44,461,450 clean reads were obtained and used for further analysis, 2,673,840 unannotated reads were retained for potential novel miRNAs prediction (Table 1). These unannotated sequences were submitted to Mireap v0.2 software to analyze their MFE (Minimum free energy), precursor structure, and Dicer cleavage site (Materials and methods). Although Stem-loop hairpin structures were considered typical characteristics for mature miRNAs, these structures are not a unique feature of miRNAs, many random inverted repeats can also fold into dysfunctional hairpins (termed pseudo-hairpins) (Zhang et al., 2006). So, it is extremely challenging to define the novel miRNAs based only on their stem-loop hairpin structures. In this study, RNAfold software (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) was used to predict the hairpins structure according to the reported criteria (Brown and Sanseau, 2005): mature miRNAs are present in one arm of the hairpin precursors, which lack large internal loops or bulges, the secondary structures of the hairpins are steady, with a free energy of hybridization lower than −20 kcal/mol, and hairpins are located in intergenic regions or introns. The potential novel miRNA candidates should meet these three criteria and form perfect stem-loop structures. Finally, all remaining novel miRNA candidates were subjected to MiPred (http://www.bioinf.seu.edu.cn/miRNA/) to filter out pseudo-pre-miRNAs using the following settings: Minimum free energy >−20 kcal/mol or p-value >0.05 (Jiang et al., 2007). The use of three miRNA precursor structure prediction programs ensured the reliability of novel miRNAs candidates. However, biological experiments should also be done to further validate their authenticity.
MicroRNAs regulate gene expression through recognizing target sites. The majority of miRNA target sites lie within the 3′ UTR of the targeted mRNA (Fabian et al., 2010). In animal genomes, it is difficult to predict target sites because the targets of miRNAs generally only display partial complementarity to the mature miRNA sequence (Millar and Waterhouse, 2005; Carthew, 2006). Because no 3′-UTR database is currently available, Mireap software was used to predict the putative target genes for those 50 novel miRNAs by searching the goat EST database at NCBI (http://www.ncbi.nlm.nih.gov/sites/entrez?db=nucest&cmd=Search&dopt=DocSum&term=txid9925%5BOrganism%3Anoexp%5D). GO is an international standardized classification system for gene annotations that provides insight into the molecular functions of genes in various biological processes. Therefore, to further understand the physiological functions and biology processes involved by these miRNAs, a total of 55,448 annotated mRNA transcripts were classified and annotated in GO database. The results showed that many functions in the goat miRNAs involve biological processes, cellular component, and molecular functions, such as biological regulation, cellular process, metabolic process, binding activity, etc.. Meanwhile, because KEGG is a systematic network database to analyze complex gene functions and cellular pathways, the target genes were classified according to KEGG functional annotations to identify pathways that were actively regulated by miRNA in the lactating mammary gland of goats. The 10 most enriched possible pathways are shown in Figure 4; it appeared that the enriched miRNAs were intensively involved in not only the metabolic pathway, but also the pathways related with disease. The results illustrate some of the possible roles of the novel miRNAs in signaling pathways during mammary gland lactation progresses, these novel miRNAs may be important for identification of the regulation networks. To better understand the regulation networks between these novel miRNA and mRNAs, further analysis are warranted.
CONCLUSIONS
In this study, three small RNA libraries from lactating mammary gland tissues were constructed and sequenced using Solexa sequencing, the identification of novel miRNAs highlights the important function of low abundant and less conserved miRNAs during the physiology of specific tissues. GO and KEGG analysis of target genes indicated that novel miRNAs might function in mammary gland physiology and lactation through regulating those target genes. Our study provided further insight into the miRNA-mediated regulation of target genes.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print







