



INTRODUCTION
The meat quality of cattle is determined by intramuscular fat deposition (marbling) (Lee et al., 2007) and could be improved by functional genomic studies of genetic factors. Beef is graded according to the amount of marbling since marbling makes beef more tender, flavorful, and juicy. It is one of the main factors used to determine beef quality grade in the United States (USDA, 1989), Japan (JMGA, 1988), and Korea. Several countries identify meat quality challenges, such as marbling, meat tenderness, carcass weight, muscling, and fat cover. All of these areas must be considered to provide consumers with high-quality products. In particular, marbling refers to the appearance of white flecks or streaks of adipose tissue between the bundles of muscle fibres in bovine skeletal muscle (Harper et al., 2001). It is driven through the development of adiposes in combination with declinging muscle growth (Hocquette et al., 2010). From one point of view, marbling might be an interaction among fat development, connective tissue or blood vessels. Kokta et al. (2004) reported the inreaction between myogenic cells and adipocytes to determine the rate and extent of myogenesis and adipogenesis during animal growth. Three genes were identified as being significantly correlated with bovine skeletal muscle based on microarray data from a gene network (Reverter et al., 2006). Jiang et al. (2009) reported that the genetic network was associated with 19 economically important beef traits. Recently, candidate genes for the marbling trait and their relationships were identified from the protein-protein interaction networks (Lim et al., 2011). Kim et al. (2011) identified the relationship between the expression of heat shock protein ╬▓1 (HSPB1) and its regulator genes from the gene network analysis in intramuscular fat of Hanwoo (Kim et al., 2011). These results reflect the fact that many biological pathways or interactions occur between muscle and fat within the skeletal muscle. Therefore, the study of marbling differences needs to analyze the complex interactions between biological pathways or genes from the network level.
Gene expression data have been used to successfully identify relationships between genes involved in biological mechanisms and to predict targetable genetic components associated with complex traits or disease states. Several studies have also shown that mRNA levels of candidate genes are heritable, affecting genetic analysis (Brem et al., 2002; Wayne and McIntyre, 2002; Schadt et al., 2003). Many complex traits in animals, such as disease susceptibility, development, and agricultural product quality, are controlled by interactions among several genes combined with environmental influences. Furthermore, patterns of covariation in the expression of multiple loci can be used to build networks that show relationships between genes and functional traits. These networks provide information on the genetic control of complex traits and can help identify causal genes that affect gene function, rather than gene expression (Haley et al., 2006). System-oriented approaches using gene expression data have been applied by animal geneticists to investigate livestock traits (Nobis et al., 2003; Donaldson et al., 2005; Smith et al., 2007), resulting in the identification and characterization of economically important causal trans-acting genes within QTL regions. These trans-acting regions share a common biological function (e.g., similar gene ontology function, metabolic pathway, transcriptional co-regulation) (Schadt et al., 2003; Gibson and Weir, 2005; Subramanian et al., 2005).
A weighted gene co-expression network is a gene correlation network created from expression profiling, with each gene having several neighbors (Peter and Steve, 2008). Gene co-expression network (GCN) is useful for identifying genes that control quantitative phenotypes and has been used as a ŌĆ£primary screenŌĆØ, to identify novel genes related to traits from thousands of possible genes. Gene expression networks serve as an effective approach for finding hub genes that have key regulatory roles. Fuller et al. (2007) demonstrated that two types of gene co-expression network analysis can find a body-weight-related gene from weighted gene co-expression network analysis (WGCNA). WGCNA analysis is applied in several research fields such as diseases (Ghazalpour et al., 2006; Miller et al., 2008), complex traits (Ghazalpour et al., 2006) and specific tissues (Oldham et al., 2006; Dewey et al., 2011).
In this study, we reported the gene co-expression network analysis of marbling trait-related genes in m. longissimus with divergent marbling phenotypes, and suggest evidences for the biological significance of highly connected genes in Hanwoo (Korean cattle).
MATERIALS AND METHODS
Microarray data processing
We used microarray experiments from intramuscular muscle samples of Korean Cattle (Hanwoo) in our previous study, related to the beef marbling study (Lee et al., 2010). Briefly, ten steers each from a low-marbled group (7.4┬▒2.4%) and a high-marbled group (23.7┬▒5.6%) were used in this study (Table 1). All arrays were processed to determine the robust multiarray average (RMA) (Irizarry et al., 2003) using the ŌĆ£affyŌĆØ software package (Gautier et al., 2004). Expression values were computed in detail from raw CEL files by applying the RMA model of probe-specific correction for perfect-match probes. These corrected probe values were then subjected to quantile normalization, and a median polish was applied to compute one expression measure from all probe values. Resulting RMA expression values were log2-transformed.
Weighted gene co-expression network analysis
We selected the 4,000 most varying probes for the generation of a weighted gene co-expression network. We calculated correlations between the gene expression profiles of each pair of genes using PearsonŌĆÖs correlation coefficients (denoted as r). Then, the correlation measures were transformed into a connection strength using power adjacency function. The power adjacency function c = |cor(xi,xj)╬▓ was used to construct a weighted network as the connection strength between two genes. The weighted network represented ŌĆ£softŌĆØ thresholding that weighed each connection as a continuous number [0, 1]. We selected a soft threshold beta (╬▓) = 18 according to scale free topology criterion. A major advantage of weighted networks is that highly robust results are obtained with regard to the choice of the parameter beta (╬▓). A major aim of co-expression network analysis is to determine subsets of nodes (modules) that are tightly connected to each other. To organize genes into modules, we used a module identification method based on a topological overlap dissimilarity measure (Ravasz et al., 2002) in conjunction with a clustering method, which detected biologically meaningful modules. The topological overlap of two nodes refers to their relative interconnectedness. The topological overlap matrix (TOM) ╬® = [Žēij] provides a similarity measure, which has proven useful in biological networks (Ye and Godzik, 2004), where lij = Ōłæuaiua and ki = Ōłæua is the node connectivity as follows:
In the case of our network, equals the number of nodes to which both i and j are connected. To identify modules, we used TOM-based dissimilarity
d ij w ( d ij w = 1 ŌłÆ Žē ij )
Connectivity and module membership
A weighted gene co-expression network identified gene modules for biological significance. Because gene modules may correspond to biological pathways, focusing the analysis on modules (and their highly connected intramodular hub genes) amounts to a biologically meaningful data reduction scheme. Highly correlated module genes are represented and summarized by their first principal component (which is referred to as the module eigengene (ME)). The ME isused to define measures of module membership (MM) which quantify how close a gene is to a given module. MM measures allow one to annotate all genes on the array and to screen for disease related intramodular hub genes. We used the intramodular connectivity Kq(i) that is biologically more meaningful than the whole network connectivity (Saris et al., 2009). It is calculated from the sum of connection strengths between a particular gene and all other genes in the module Kq(i) = ŌłæjŌłłq,jŌēĀ1|Cor(xi, xj)|╬▓, where q denotes a specific module. We also used the MMq(i,) which is the correlation of the ME and the gene expression profile. As explained in detail in (Horvath and Dong, 2008), the MM of gene i in module q can be defined MMq(i) = Cor(xi, MEq), where larger absolute values mean greater similarity between a gene xi and the q-th module eigengene. The statistical significance of MM (denoted as p MM red) is carried out from the correlation test p-value of the WGCNA package. Finally, we can identify genes that have a high significance for marbling score as well as high MM in interesting modules using the gene significance (GS) and MM measures (Peter and Steve, 2008). We first defined a measure of GS that is obtained from the correlation between the gene and the trait. The higher the i-th geneŌĆÖs |GS(i)|, the greater its biological significance. For the i-th genes, we identified GS for marbling score (denoted as GS marbling score) as the absolute value of the Student t-test statistic for testing differential expression between high- and low-marbled groups. We defined a measure of module significance (denoted as p.MM.red) as the eigengene significance that is the correlation between the ME and the expression profiles.
Functional enrichment analysis
We performed functional enrichment analysis in given modules that were associated with marbling score enrichment in the Gene Ontology or KEGG pathway terms, using the Database for Annotation Visualization and Integrated Discovery (DAVID) tool (http://david.abcc.ncifcrf.gov/). It computes a fisherŌĆÖs exact test p-value. Functional relationships of our genes of interest were used in the Pathway studio program (Stratagene, La Jolla, CA, USA) (Nikitin et al., 2003). We investigated the common regulators and targets of the significant genes in the modules.
RESULTS AND DISCUSSION
Weighted gene co-expression network analysis
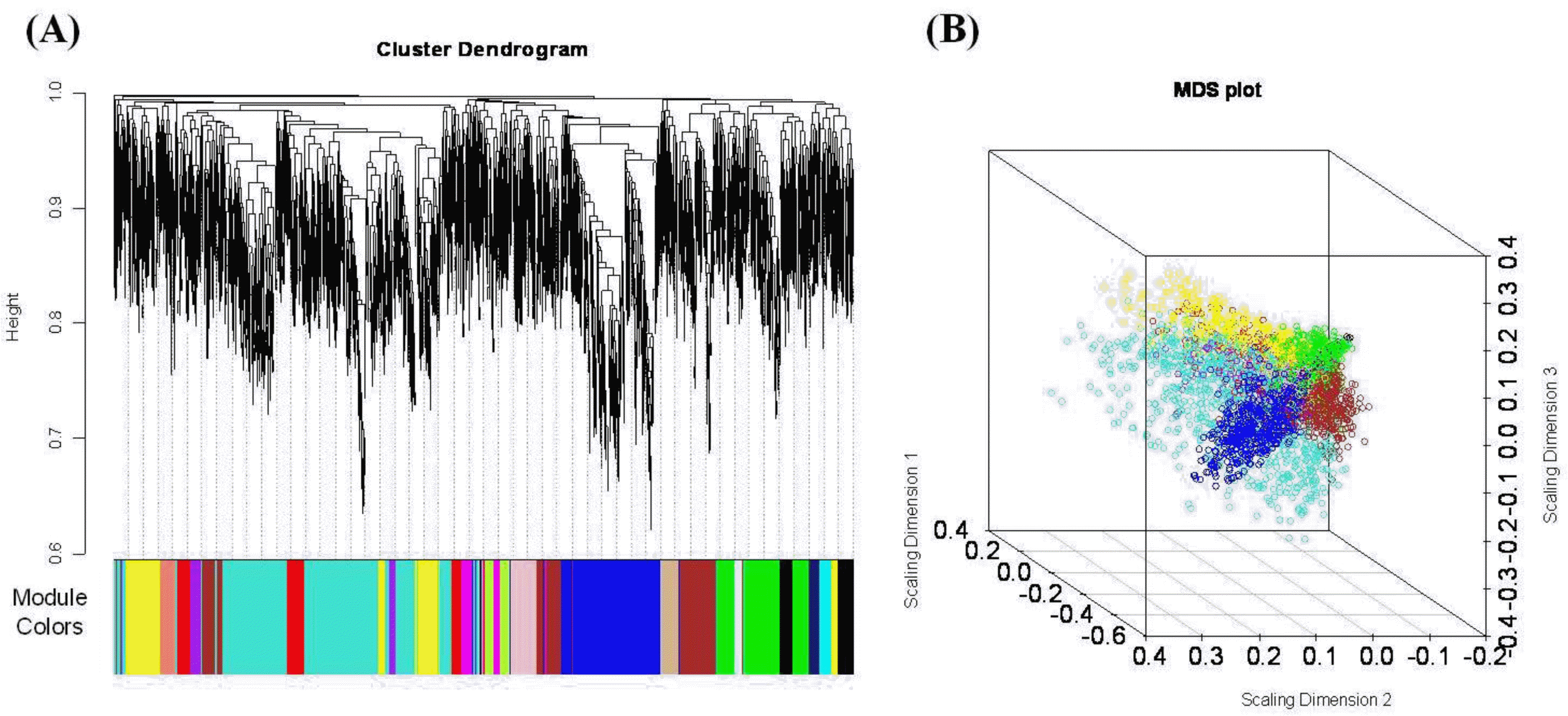
We used WGCNA in a first attempt to identify marbling score associated coexpression modules and their key functions. A weighted gene co-expression network was constructed using expression data from the high- and low marbled groups, utilizing the 4,000 most varying transcripts from the 24,128 transcripts present on the array. To find modules of highly correlated genes, we used average linkage hierarchical clustering, which uses the TOM as dissimilarity. We were able to identify 17 distinct modules (except for the ŌĆ£greyŌĆØ module, which is not grouped into any module) for groups of genes with high topological overlap. Figure 1 shows the co-expression modules ranging in size from 41 (lightcyan) to 1,024 (turquoise) genes. The mean overall connectivity is 24.6, and ranged from 8.83 (midnightblue) to 34.44 (turquoise). Detailed information about all genes and their network properties are calculated (data not shown).
Detection of co-expression modules related to marbling score
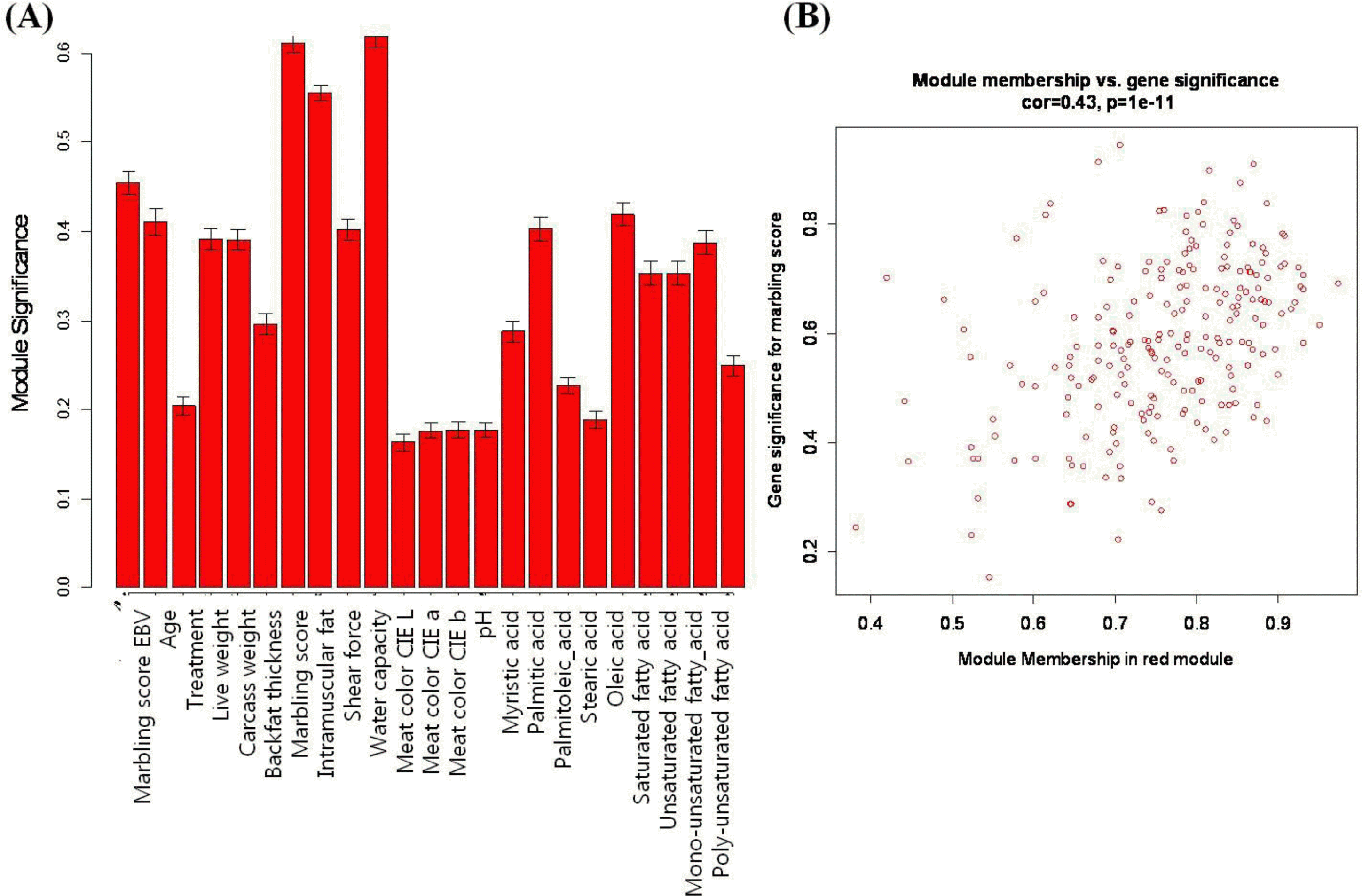
The coexpression modules correspond to branches and are color-coded (black, blue, brown, cyan, green, greenyellow, lightcyan, magenta, midnightblue, pink, purple, red, salmon, tan, turquoise, and yellow module). We identify modules that are significantly associated with the measured phenotypic traits. We found that the module significance measures in the three modules (red, tan and lightcyan) were significantly correlated (Supplementary data 1). The red module (referred as MEred) was the most significant (correlation with marbling score r = 0.77, correlation p = 0.008) for marbling score. It also showed significant results for intramuscular fat (r = 0.72, p = 0.02) and water capacity (r = 0.79, p = 0.006). Figure 2(A) shows red module significance against all traits. The tan module (referred to as MEtan) is significantly associated with three phenotypic traits: marbling score (r = 0.68, p = 0.03), intramuscular fat (r = 0.74, p = 0.01) and meat color CIE L (r = 0.62, p = 0.05). The red and tan modules were related to marbling score, intramuscular fat. Generally, intramuscular fat is often called an indicator of marbling, because they are highly correlated. The genetic and phenotypic correlations between them were 0.69 to 0.74 and 0.7, respectively (Park et al., 1994; Crews et al., 2003). The lightcyan module is related only to marbling score (r = 0.66, p = 0.04). We also investigated the relationship of the MEs to other phenotypic variables. Table 2 shows the modules that have significant p-values against the types of phenotypes.
As detailed in the Methods section, we calculated a measure of MM that can define each module. Large absolute values of MEred(i), MEtan(i) or MElightcyan(i) indicate the gene is closed to the red, tan or lightcyan module. In contrast, if MMred(i) is closed to 0, then ith gene is uncorrelated with the red module eigengene and is unlikely to be part of the red module. We also quantify the association of individual genes with the marbling score trait in each module by determining GS as the absolute value of the correlation between the gene and the trait. Figure 2(B) shows a relationship between the GS and MM in the red module (r = 0.43, p = 1e-11). However, there is no significant result (r = 0.079, p = 0.44) between GS and MM in the tan module. This implies that hub genes of the red module also tend to be highly correlated with marbling score. We reported 84, 17 and 2 probes that have significant results (pŌēż0.05) with the GS and the MM against the marbling score in the red, tan and lightcyan module, respectively. Network properties of the top-ranking genes are shown in Table 3. For example, glomulin, FKBP associated protein (GLMN), showed the most significant result for marbling score (r = 0.95, p.GS.marbling score = 3.69e-5) in the red module. This is involved in differentiation of vascular smooth muscle cells (VSMC) (McIntyre et al., 2004) and indicated as a marker of VSMC. According to Davies et al. (2005), the generation of lipid-filled VSMC resulted from either adipocyte differentiation or direct promotion of lipogenesis as the result of LXR/SREBP1c activation in humans (Davies et al., 2005). Neuregulin 1 (NRG1), integrin-binding sialoprotein (IBSP) and solute carrier family 46, member 1 (SLC46A1) have the largest module membership (MM = 0.93) in the red module. These genes also show significant p-values for gene significance against the marbling score phenotype.
Pathway and GO analysis for the red module
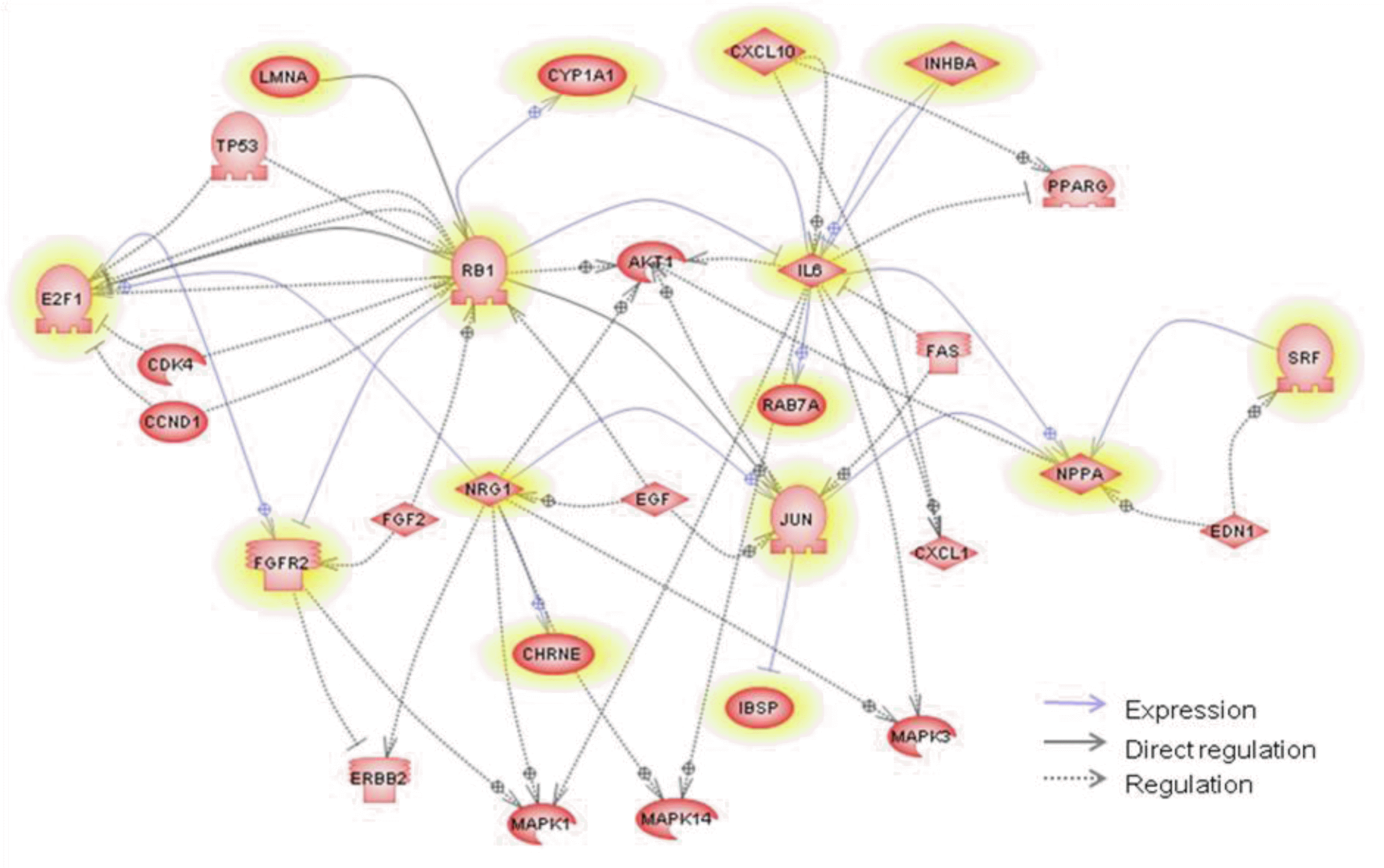
We performed functional enrichment analysis for the red module according to the GS and MM measurement. GO and biological pathway analysis were used to search for the biological significance or functional relationship of the significant genes associated with marbling score. We explored the functional relationship (expression, regulation and direct interaction) in the red module using the pathway studio program. Out of 15 pathway annotated genes, 8 muscle-related genes (NRG1, RB1, JUN, CHRNE, CXCL10, IL6, SRF and FGFR2) have a direct relationship in the pathway analysis (Figure 3). These genes have significant p-value (p<0.05) for MM or GS in red module for marbling score. The NRG family have been observed to stimulate myotube formation and muscle specific gene expression (Florini et al., 1996; Lebrasseur et al., 2003) and facilitate glucose uptake that is an important factor for improving marbling in adipocyte of beef cattle (Suarez et al., 2001). Activation of NRG/ErbB signaling may also mediate one or more adaptive growth and metabolic responses of skeletal muscle to exercise. Fibroblast growth factor receptor 2 (FGFR2) is a member of four transmembrane tyrosine kinase receptors and affects skeletal muscle myogenesis (Rhoads et al., 2009). The function of satellite cells during muscle regeneration is regulated by many growth factors and cytokines such as fibroblast growth factor (FGF) and transforming growth factor-╬▓ (TGF-╬▓) families, insulin-like growth factors-1 and -2 (IGF-1, IGF-2), hepatocyte growth factor (HGF), and interleukin-6 (IL-6) (Grefte et al., 2007). One of the FGF family, polymorphisms in the FGF8 is associated with carcass quality, growth and feed efficiency in beef cattle (Moore and Marques, 2008). Interleukine 6 (IL6) regulates skeletal muscle differentiation and metabolism. In particular, it increased glucose incorporation into glycogen, glucose uptake, lactate production, and fatty acid uptake and oxidation in humans (Al-Khalili et al., 2006). Retinoblastoma 1 (RB) plays an important role in determining whether myoblasts proliferate or differentiate (Rosenthal and Cheng, 1995). RB family proteins promote adipogenesis by direct interaction with C/EBPs (Chen et al., 1996). Chemokine ligand 10 (CXCL10) is differentially expressed in the longissimus tissues from Meishan, Meishan├ŚLarge White cross and Large White pigs (Li et al., 2010). Serum response factor (SRF) was shown to be differentially expressed between fat and lean and between different muscles using RT-PCR in chickens and was suggested as a potential regulator of several functional candidates affecting glycogen turnover in the muscle for meat quality (Sibut et al., 2011). Jun oncogene (JUN) is called an activator protein 1 (AP-1) and is known to inhibit myogenic differentiation (Su et al., 1991). It controls the transcription factor involved in myogenesis and those involved in cell proliferation (Li et al., 1992). AP-1 is also one of the transcription factors binding in the promoter of FABP4 with CEBP╬▒ (Shin et al., 2009). Recently, the mutation of cholinergic receptor, nicotinic, epsilon (CHREN) is significantly associated with muscle growth in beef cattle from primer-extension assay (Sevane et al., 2011). These results indicate that the genes in the red module may function in regulating muscle growth or fat-related mechanisms and co-expressed genes with similar functions in the module. In addition, we explored regulatory relationships (i.e., common regulators and targets) between 15 direct-interacted genes using pathway studio. The common targets or regulators are shown in Figure 3 with the direct interaction relationship. We found the common regulators based on an assumption that the genes within a similar biological pathway are controlled by common regulators. E2F1 is one gene of the E2F family and is a candidate to be a transcription factor controlling corticotropin releasing hormone (CRH) for marbling and subcutaneous fat depth in beef cattle (Wibowo et al., 2007). In longissimus muscle tissue expression during growth in the porcine, E2F1 also showed a significant relationship with differential expressed genes as a transcription factor with myogenin and PAX3 (DŌĆÖAndrea et al., 2011). In our network analysis, E2F1 is a member of the red module and regulates FGFR2. The FGF family plays a role in cell growth, such as cell proliferation and angiogenesis. The FGFR2 protein is induced in the mid-to-late G1 phase of the cell cycle by E2F1 (Tashiro et al., 2003).
Finally, we investigated the functional bias of the significant genes according to GO classification and understood the biological significance of the module genes, and determined the putative pathways using DAVID. Table 4 lists the significant gene ontology terms and the representative genes. Due to the incomplete annotation of the bovine genome, 168 of 222 probe sets were annotated (Table 4). In significant GO terms of Biological processes, the regulation of biological quality (GO:0065008) indicates that the process modulates a measuable attribute of an organism or part of an organism, such as size, mass, shape, color, etc. This result is also reflected in the pathway analysis. The term is included in 5 pathway hub genes (IL6, CHRNE, RB1, INHBA and NPPA) of 12 annotated genes by gene ontology. Collagen, type IX, alpha 1 (COL9A1) is detected in the other genes. Significant associations of the COL9A1 gene with body length, depth and width have previously been reported in pigs. Recently, it has also been related to logissimus muscle area from assocation analysis in the pig (Fan et al., 2009). These findings suggest that genes in the red module tend to be highly enriched with meat quality and have a potential role to change or control a specific phenotype for animal production.
CONCLUSION
A major objective of this study was to construct the gene co-expression network and then to find hub modules or genes associated with the marbling score. Therefore, we attempted to find coexpression patterns associated with marbling in Hanwoo (Korean cattle) by the WGCNA method. As a result, three large co-expression modules were significantly associated with marbling score and intramuscular fat. Among these three modules, we focused on the red module for functional enrichment analysis. This is because the tan and lightcyan modules have not shown a significant correlation between gene significance and module membership in each module. Through the pathway and gene ontology analysis, we consistently observed that hub genes within the red module were predominantly a co-expression group having biological pathways related to skeletal muscle. We noticed overlapping genes from the analysis, and five genes (IL6, CHRNE, RB1, INHBA and NPPA) belonged to a red module. These genes are shared in skeletal muscle related biological pathways that might represent a phenomenon occuring in muscle with highly divergent marbling phenotype as key drives. Our results do not point to a single biological pathway or candidate gene like a standard differential expression analysis. Instead, we find several highly significant biological pathways and patterns of co-expressed genes as key drivers in the marbling score related modules. These results will provide valuable information for the additional biological study of meat quality in Hanwoo (Korean cattle).
Supplementary data 1.
The relationship between a module and a trait. Each row corresponds to a module eigengene, column to a trait. Each cell contains the corresponding correlation and p-value. The table in color-coded by correlation according to the color legend.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print







