



INTRODUCTION
The pig (Sus scrofa) has been widely used as a model for studying the development of human organs and disease progression, owing to the similarity in size and physiology of the two species (Butler et al., 2007; Lunney, 2007; Ojeda et al., 2008). The Landrace is one of the pig breeds that are used world-wide in research and production because of its genetic stability and high lean percentage.
The alimentary tract is essential for absorption of nutrients and the function of the immune system. A notable feature of the intestine is the complex changes that occur over the lifetime of an individual (Weaver et al., 1991; Thompson et al., 2008). The development of the intestine is regulated by a large number of factors, including nutrients, the micro flora, the epithelium, innate and adaptive immunity (Pacha, 2000; Caicedo et al., 2005; Donovan, 2006; Commare et al., 2007; Ojeda et al., 2008), which constitute the intestinal ecosystem. It is known that the adaptive change involved in enteric development is highly conserved and is a complex process (Gilbert and Lloyd, 2000). The underlying molecular mechanism of intestinal development is associated with genes and the corresponding proteins that participate in this change, and with the interactions and signal transduction processes of the gene and/or protein networks. It is almost impossible. However, to understand the complex interactions of genes and proteins by studying them in an individual and static manner. The study of gene regulatory networks makes it feasible to analyze the interactions of genes in an integral and dynamic way (Tomita et al., 1999; Gilbert and Lloyd, 2000). Gene regulatory networks have attracted considerable research interest owing to the rapid accumulation of genomic information (Kitano, 202). Comprehensive assessment of the gene expression profiles needs to be carried out in order to identify gene networks (Kitano, 2002), and the high-throughput techniques of molecular biology can be used to provide insight into such networks.
Microarray technologies have enabled researchers to explore the dynamics of transcription on a genome-wide scale (Young, 2000; Jiang and Deyholos, 2006; Zhang et al., 2008), and they provide a systematic insight into the crucial factors that control development. They can be used to elucidate the mechanisms of intestinal development at a molecular level. Studies of gene expression profiling using microarray technologies in the development of plants and the human fetal pancreas have made significant progress (Sarkar et al., 2008; Wechter et al., 2008), but there have been no reports of such studies on the development of the porcine intestine. In the present study, we aimed using microarray technology to identify the significant pathways and key genes that regulate the intestinal development of Landrace piglets.
MATERIALS AND METHODS
Animals and tissue preparation
All animals used in this study were managed humanely according to the established guidelines of the Animal Care Committee, Sichuan Agricultural University, P. R. China. The four Landrace sows were mated with a boar of the same breed. Two male piglets were sacrificed at 0, 3, 8, 14 and 21 d after birth, respectively. The mid-section of the jejunum of each piglet was dissected rapidly. The samples were snap-frozen in liquid nitrogen and stored at ŌłÆ80┬░C until further use.
Microarray analysis
Isolation of RNA was performed with the QIAGEN RNeasy mini kit (QIAGEN, Valencia, CA, USA) following the manufacturerŌĆÖs instructions. The RNA samples were then analyzed using the Affymetrix GeneChip┬« Porcine Genome Array (Affymetrix, Santa Clara, CA, USA), which contains 23,937 probes sets that interrogate approximately 23,256 transcripts from 20,201 genes of S. scrofa. Statistics were computed using the GeneChip Operating Software (GCOS). The data discussed in this article have been deposited in the NCBIs Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO Series accession number GSE13456.
Time sequence profile analysis of gene expression
The differentially expressed genes from four time points (d 3, 8, 14, and 21) were compared with those of d 0 using Affymetrix GCOS software. The raw values for gene expression were converted to log2ratio. According to the analysis strategy for an experiment representing a cluster of short time series (Ernst et al., 2005), 80 profiles were defined in the experiment. The profiles were analyzed that had the most significant probability assigned in comparison with that expected by the Fisher exact test and the multiple comparison test. If the p-value of the profiles was less than 0.01, the gene expression profiles were identified to be significantly different with respect to time.
Functional analysis of significant profiles of co-expressed genes
According to previous studies (Yu et al., 2003; Pan et al., 2008; Rawat et al., 2008), co-expressed genes have similar functions. The important functions were analyzed for those profiles found to be significant using Gene Ontology (GO), which organizes genes into hierarchical categories based on biological processes, molecular functions, and cellular components (Ashburner et al., 2000). To undertake GO analysis, the p-values from the FisherŌĆÖs exact test for over-representation of the selected genes in all GO biological categories are computed, as described previously. Significance of the GO was accepted when the p-value was less than 0.05. Functional annotation can provide a detailed description of the roles of genes located in deeper categories of GO, according to the hierarchical organization of GO. Therefore, the enrichment value was defined to filter the deeper categories of GO: the higher the enrichment value of GO, the more target GO were expected. Within a given category, the enrichment Re is given by equation,
where nf is the number of differentially expressed genes within the particular category, n is the total number of genes within the same category, Nf is the number of different genes on the entire microarray, and N is the total number of genes on the microarray.
Pathway analysis of differentially expressed genes
A pathway is defined as a functional unit in which genes interact with each other and achieve a process of signal transduction. To explain test phenotypic features with respect to development processes, significant pathways are identified from differentially expressed genes. This is based on the p-values of the FisherŌĆÖs exact test for over-representation of the selected genes in all pathway categories, as described perviously (Shalgi et al., 2007). A significant pathway was specified when the p-value was less than 0.05. To assess the significance of a particular category with respect to random chance, the false discovery rate (FDR) was estimated for a set of categories. After resampling 5,000 times, the FDR was defined by equation, where Nk refers to the number of FisherŌĆÖs test p-values less
than ├Ś2 test p-values.
The p-values obtained using FisherŌĆÖs exact test are lower than ├Ś2 test p-values. Pathway enrichment analysis was performed according to a method reported previously (Kanehisa and Goto, 2000) to investigate whether particular functions were enriched among the genes that control distinctive characters in groups of genes that were differentially expressed on d 3, 8, 14, and 21, in comparison with d 0 (the control time point). With increasing enrichment, the corresponding function becomes more significant. Within a given category, the enrichment Re is given by equation, where nf is the number of different genes within the particular pathway category, and n is the total number of genes within the same category, Nf is the number of different genes on the entire microarray, and N is the total number of genes on the microarray.
Construction and topological analysis of the gene co-expression network
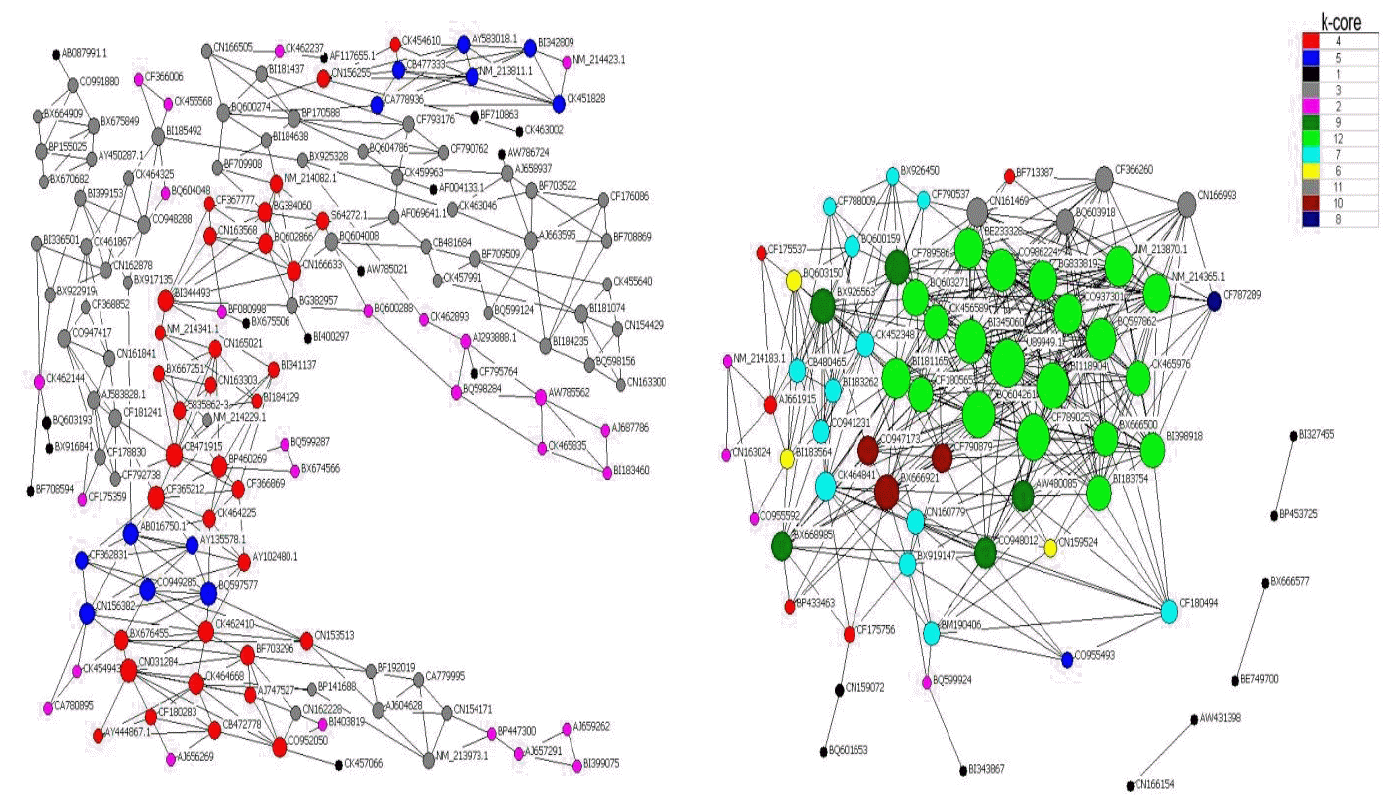
We transformed the normalized expression values of profile 66 and profile 13 from the individual correlation analysis into measures of pair-wise connection strengths for the construction of a co-expression network (Carlson et al., 2006). The network edges were specified to be more than 0.9, which is the value of the coefficient correlation that incidates a strong relationship, such as co-expression of genes. The centrality of a network is represented by the central degree (Barabasi and Oltvai, 2004). It is possible to determine the characteristic variables that are related to the distances among genes. The core of maximum order is referred to as the main core, or the highest k-core of the graph (Huber et al., 2007). A k-core subgraph of a graph can be generated by deleting the vertices recursively from the graph whose degree is less than k (Huber et al., 2007). Larger values of ŌĆ£corenessŌĆØ clearly correspond to vertices of larger degree and a more central position in the structure of the network. In this work, we applied the notion of the k-core subgraph to predict similarity of gene function (Altaf-Ul-Amin et al., 2006). The nodes labeled by the same color may represent similar gene ontology terms. The highlight ontology of nodes of the same color was assessed by counting the genes of the same GO that had the same color.
Real-time quantitative PCR
Fourteen differentially expressed genes (AREG (Amphiregulin), ATRN (Attractin), CD59 (CD59 molecule), CLU (Clusterin), CST3 (Cystatin C), DPEP1 (Dipeptidase), EPHX1 (Epoxide hydrolase), LEAP2 (Liver expressed antimicrobial peptide 2), LOC396871 (Arginine rich antibacterial peptides), MS4A2 (Membrane-spanning 4-domains), PIAP (Putative inhibitor of apoptosis), RETN (Resistin), UBP (Ubiquitin-specific protease), FBP (Folate binding protein) dentified in the array experiment were selected and analyzed by RT-qPCR. Aliquots of the same RNA samples used for the microarray analysis were used for the real-time PCR. The reverse transcriptions (RT) were carried out with the total cellar RNA using TRIZOL Reagent┬« (Invitrogen California, USA). Each sample of cDNA was diluted 1:4 fold in sterile ddH2O, and 1 ╬╝l of this dilution was used as the template for qPCR. Primers for the PCR reactions were designed by DNAStar (DNASTAR Inc) to have a Tm of 62 to 65┬░C and an optimal annealing temperature of 63┬░C, with a length of 100 and 250 bp. Real-time PCR was performed with SYBR┬« Premix Ex TaqTM (TaKaRa, Dalian, China) in 20 ╬╝l reaction volumes using the iCycler System (BioRad, Richmond, CA, USA) according to the manufacturerŌĆÖs instructions. Each PCR reaction contained 1 ╬╝l of cDNA, 0.2 ╬╝M of each of the primers (Table 1) and 10 ╬╝l of PCR master mix. The initial denaturing time was 95┬░C per minute, followed by 40 cycles consisting of 94┬░C for 15 s, 63┬░C for 20 s, and 72┬░C for 30 s with a single measurement of fluorescence.
RESULTS
Microarray analysis
To characterize the genes involved in intestinal development during the first 21 d in pigletsŌĆÖ life, total RNA was extracted from the mid-section of the jejunum on postnatal d 0, 3, 8, 14 and 21. Gene expression was analyzed using a porcine genome array and statistics were computed using GCOS. In total, 8,427 significant transcripts were identified. Among these statistically significant expression ratios, probes representing 4,789 unique genes increased in transcript abundance at least 2.0 fold at one or more time points, while probes representing 3,618 unique genes decreased in abundance at least 2.0 fold. Thus, at least 24% of the total number of transcripts from S. scrofa were strongly (i.e. >2.0 fold) induced by intestinal development; however, at least 18% of transcripts were repressed by an equivalent amount (Table 2).
Gene expression profiles significant to the development process
During the course of intestinal development from d 0 to 21, 80 model profiles were identified in which we defined the value of maximum unit change to be 1. For these model profiles, 30 were identified as significant (Figure 1). Among them, the two most significant profiles were profile 66 and profile 13, according to ascending order of the p-values (Figure 2). If the p-value of a profile was less than 0.01, the gene profile was identified as one that changed significantly with time. According to this principle, the lowest p-value profiles may represent the regulatory genes that affect the development of the porcine 539 genes, the expression of which increased significantly at d 3 and maintained a similar level of expression until d 21. In contrast, profile 13 had a significance level of 8.8├Ś10ŌłÆ87 and contained 351 genes, the expression of which decreased significantly at d 3 and maintained a similar level of expression until d 21.
Significant gene functions and genes of significant profiles
Results of the analysis of GO categories for profile 66 implied that the main enriched GO categories were focused on control checkpoints for cell size, cell wall biogenesis, deoxyribonucleotide catabolic processes, fungal-type cell wall biogenesis, fungal-type cell wall organization and biogenesis, lymphocyte chemotaxis, M phase-specific microtubule processes, membrane depolarization, microvillus biogenesis, mitochondrial depolarization, mitotic metaphase, etc. during growth from 0 to 21 d (Table 3). To further investigate the most significant functions, the annotation of the GO was analyzed using Amigo (a website tool (http://amigo.geneontology.org/cgi-bin/amigo/go.cgi). It was concluded that the process of the cell cycle initiated at a checkpoint and the immune response to fungi stimulated by fungal infection were the most significant phenomena that occurred from d 3 to d 21.
In contrast, the main enriched GO categories for profile 13 were focused on folic acid transport, negative regulation of endocytosis, the phosphatidylcholine biosynthetic process, phosphatidylcholine metabolic processes, the response to cobalt, manganese and zinc ions, catabolic processes for aminoglycans, glycosaminoglycans, and leucine, leucine metabolic processes, a lipopolysaccharide-mediated signaling pathway, Mo-molybdopterin cofactor biosynthetic processes, and Mo-molybdopterin cofactor metabolic processes, etc., as presented in Table 4.
Distribution of significant pathways of differentially expressed genes
The pathway was analyzed using the KEGG and Genmapp databases. The significant pathways for differentially expressed genes were applied to the Fisher exact test and tests of multiple comparisons to obtain the significant functions. From the results of Table 6, it can be seen that the metabolism of xenobiotics by cytochrome P450, AlzheimerŌĆÖs disease, antigen processing and presentation, the PPAR signaling pathway, complement and coagulation cascades, cell adhesion molecules (CAMs), autoimmune thyroid disease, allograft rejection, graft-versus-host disease, natural killer cell mediated cytotoxicity, and cytokine-cytokine receptor interaction were the significant pathway categories. It is well known that owing to the changes in diet and living environment that occur after the birth of piglets, adaptive changes occur in the internal environment. In this process various immune reactions are activated, such as antigen processing, complement cascades, allograft rejection, graft-versus-host disease, natural killer cell mediated cytotoxicity and cytokine-cytokine receptor interaction. It can be concluded. Therefore, that the immune response may be coupled with the growth of the organism (Table 5).
The gene co-expression network
The gene co-expression network was constructed with respect to gene function associations. Given that the elements of a network represent a variety of gene regulatory abilities, a large-scale gene network can be divided into subgraphs, named k-core networks, in which all genes affect each other (Huber et al., 2007). Generally, the complexity of gene relationships increases with the rank of the k-core value. In the light of the definition of the k-core network, the core status within a large-scale gene network consists of subgraphs of higher k value. In this work, we used the k-cores of protein-protein interaction networks to define the main gene functions in the main subgraph. Moreover, we aimed to find the main GO assigned by the maximum number of genes in a separate k-core and then to define the key gene functions at each level of the network.
It has been reported that the core functions at the core status of a network have a top k-core level (Altaf-Ul-Amin et al., 2006), and a higher degree but lower clustering coefficient (Barabasi and Oltvai, 2004). A k-core subnetwork with a k-value of 12 was located at the core status within the large scale gene network (Figure 3). Using the hierarchical categories of gene ontology, it could be concluded that most of the genes in this subnetwork are attributed to cell communication, which may be related to cell division or proliferation through cell adhesion and transport of nutrients (for details of the genes, Tables 3 and 4). In addition, most genes in the secondary network (with a k-core value of 11) play an important role in metabolic processes of nitrogen compounds and amines (for detailed information, Tables 3 and 4), except for those genes without detailed functional annotation. Among these genes, CN161469 is similar to methylcrotonoyl-Coenzyme A carboxylase 2 (beta) when analyzed by BLAST, and it takes part in the regulation of other genes within the secondary level subnetwork because of the higher value of its degree. Most notably, the gene U89949.1, which encodes a folate binding protein, possessed the highest k-core and degree, and the lowest clustering coefficient (Table 6). These features indicate that this is the core gene that is located at the center of both the large scale network and the 12 k-core subnetwork, and it regulates directly 28 neighboring genes.
Validation of the gene chip data by RT-PCR
To confirm whether the genes identified were differentially expressed, quantitative RT-PCR analysis for specific transcripts was carried out as described previously. Based on our microarray analysis, 14 genes, including three antimicrobial peptides (LEAP2, LOC396871, and MS4A2), two proteins involved in the regulation of proliferation and apoptosis of epithelial cells (PIAP and AREG), and genes associated with a number of physiological and metabolic processes (ATRN, CD59, CLU, DPEP1, EPHX1, RETN and UBP) were selected. The core gene FBP was also involved in the RT-PCR analysis. For all of these genes, the expression ratios measured by RT-qPCR and by microarray were highly correlated (r = 0.74, 0.73, 0.95, 0.93 at d 3, 8, 14, 21 respectively) (Table 7). Genes involved in apoptosis, signal transduction, and energy and protein metabolism, such as CLU, MS4A2 and UBP, were up-regulated during suckling, as indicated by both the Genechip and RT-qPCR experiments. However, HCRTR2 displayed significant differential expression when detected by RT-qPCR but no significance on Genechip testing. The correlation of the results from the RT-qPCR and Genechip testing helped us to analyze the reliability of the assays so as to reveal true differences in the gene expression profiles in the intestine.
DISCUSSION
Owing to the continuous improvement of microarray technology, the rapid development of bioinformatics and the depth of research on genomics and proteomics, global genome expression profile analysis provides a more comprehensive and dynamic analysis of intestinal development at the genome-wide transcriptional level (Ravasz et al., 2002; Schlitt et al., 2003; Stears et al., 2003; Barabasi and Oltvai, 2004; Han, 2008; Lee et al., 2008). The understanding and description of cells, organs and living individuals on a system level is a new area of biological research (Davidson et al., 2002; Kitano, 2002; Oltvai and Barabasi, 2002). Various kinds of model organism have been proved to be useful in this area (Covington et al., 2008; Sarkar et al., 2008; Schweikl et al., 2008; Wechter et al., 2008). To our knowledge, this is the first study involving whole genome analysis of the genes related to intestinal development in Landrace piglets at different stages of growth (0, 3, 8, 14, and 21 d after birth).
In the present study, more than 8,000 differentially expressed transcripts were identified by microarray technology. This technique was validated by RT-qPCR for utilization in a comprehensive study of gene expression profiles. However, many of these transcripts had no detailed information in the cDNA resources deposited for swine, which limited the analysis of their biological importance. This indicates that many porcine genes have yet to be analyzed. Complex molecular events pertinent to intestinal development occur during the suckling stage in piglets (Hall and Byrne, 1989; Buddington, 1994; Thaler and Cummings, 2008). We analyzed the development process of the gene expression profile, and 80 model profiles were obtained in all. These profiles do not cover all the possible profiles that could be generated in the intestine of Landrace piglets during suckling. Among these profiles, 30 were identified as significant and two of them (profiles 66 and 13) were highly significant. Significant profiles may indicate that common functions were mainly attributed to the co-expressed genes, and these functions influence the biological characteristics of the organism (Gracey et al., 2004). We can therefore conclude, according to the significance of the profiles, that the genes assigned to these two profiles may be highly specific for the intestinal development of piglets in early life. In order to obtain a better understanding of the genes assigned to these two profiles and to elucidate the molecular mechanism of development of the intestine, analysis of GO categories, pathway analysis, and topological analysis of the gene co-expression network were carried out.
Annotation of GO categories has proved to be remarkably useful for the mining of functional and biological significance from the results of microarray studies. In our study, significant GO categories were determined by the hypergeometric distribution and multiple comparison tests. The results obtained for the GO categories implied that the genes in profiles 66 and 13 were involved in many biological processes, among which the distinguished phenomena were initiation of the cell cycle at a checkpoint and the immune response to fungi for profile 66, while the folic acid transport process was most notable for profile 13. These results suggest that the genes in profile 66 may regulate cell proliferation and differentiation, and the genes in profile 13 may modulate the metabolism of nutrients.
Analysis of the pathway for the lists of differentially expressed genes may allow us to find the target pathway that regulates phenotypic differences. Pathway analysis in the present study indicated that the genes in profiles 66 and 13 focused mainly on the immune system, metabolism, cell adhesion molecules, etc. It may be concluded that these genes play an important role in the above pathways, which agrees with the results obtained from the analysis of the GO categories.
During biological processes, a macromolecular network can be constructed according to the yeast two-hybrid method (Y-2H) (Smidtas et al., 2006) or an algorithmic prediction based on the gene function correlation and expression profiles (Nikiforova and Willmitzer, 2007). The complexity of the network model based on the high throughput gene expression test using the algorithmic prediction made it feasible to determine snapshots of protein-protein interactions, gene expression regulatory networks, and metabolic networks among different gene groups (Carlson et al., 2006). To find the most effective genes in profiles 66 and 13, the gene co-expression network was constructed using the algorithmic prediction, and topological analysis was carried out, the results of which demonstrated that more than 20 effective genes were identified. Most of these genes were attributed to cell communication, which may be related to cell division or proliferation through cell adhesion and the transport of nutritients. Among these genes, CN161469 was found to be similar to methylcrotonoyl-Coenzyme A carboxylase 2 (beta), which suggests that it possesses the similar biological functions. In addition, the gene U89949.1, which encodes a folate binding protein that is involved in the transport of foliate (Vallet et al., 2001; Kim and Vallet, 2004), played an major role in the network. This finding was in accordance with the results of the assessment of the GO categories and pathway analysis. It has been demonstrated that down-regulated genes reflect a state of malnutrition, which may be induced by a lack of the essential molecules for growth, such as folic acid, trace elements, and aminoglycan, in early life. In profile 13, the gene U89949.1 was down-regulated, which may imply a decrease in folic acid transport that may subsequently induce malnutrition. This gene therefore provides a channel that allows regulation of the metabolism on a molecular level to balance malnutrition and overŌĆōnutrition.
IMPLICATIONS
In order to increase our understanding of the molecular mechanisms involved in intestinal development, it is essential to obtain more information on the functions of genes in the mammalian genome and their corresponding products. In the present study, we have provided a rich new information resource, which describes the significant pathways and effective transcripts that are related intimately with porcine intestinal development. Although there was a certain degree of ambiguity when we established the regulatory network based on the expression profiling data, this represents a valuable attempt to guide further in-depth research into the candidate genes and signal transduction pathways involved in intestinal development.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print







